
Information endorsed by… ![]()
What is leukaemia, bone marrow and what are the types of blood cells?
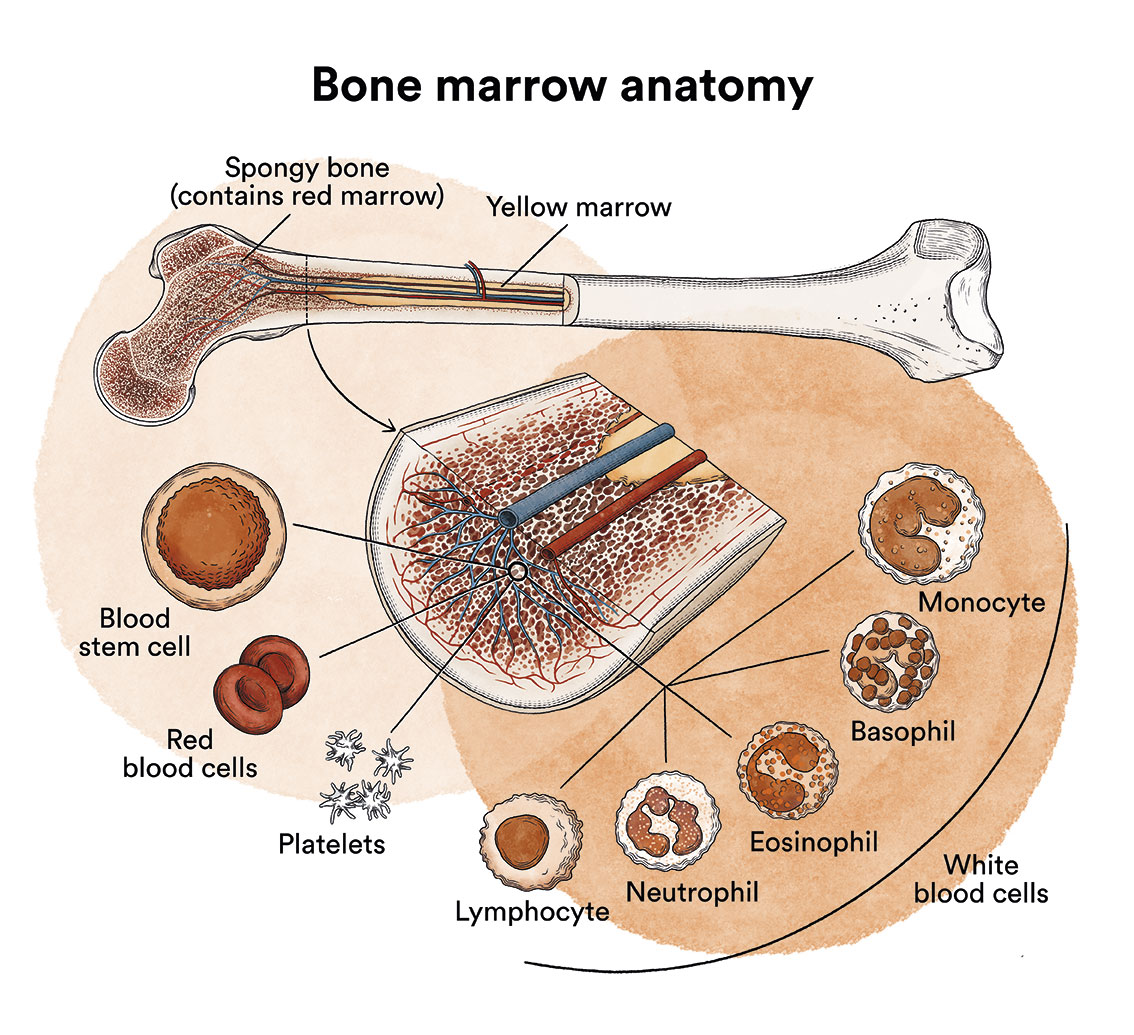
Leukaemia is a type of blood cell and bone marrow cancer. See section Leukaemia, bone marrow and blood cells.
What is juvenile myelomonocytic leukaemia?
 Juvenile myelomonocytic leukaemia (JMML) occurs when the stem cells or progenitor cells in the bone marrow , which are responsible for the generation of all blood cells, behave abnormally, proliferating uncontrollably and producing large numbers of monocytes in the blood that infiltrate various of the patient’s organs.
Juvenile myelomonocytic leukaemia (JMML) occurs when the stem cells or progenitor cells in the bone marrow , which are responsible for the generation of all blood cells, behave abnormally, proliferating uncontrollably and producing large numbers of monocytes in the blood that infiltrate various of the patient’s organs.
Juvenile myelomonocytic leukaemia is a very rare type of leukaemia, which has an incidence of only 1.2 cases per 1 million children under 14 years of age, and is more common in males (2 out of 3 patients). Between 5 and 10 cases of JMML are diagnosed every year in Spain, mostly in children under 2 years of age, although it can also occur at older ages, especially in patients with neurofibromatosis type 1. Approximately 10% of cases of JMML occur in infants under the age of 3 months.
What are the causes of juvenile myelomonocytic leukaemia?
 The specific causes of juvenile myelomonocytic leukaemia are not known. However, five genes have been identified as mutated in the bone marrow cells of 90% of JMML patients. These mutations are considered to be the triggers of the disease. In the remaining 10% of patients the genetic mutation that acts as a leukaemia promoter is still unknown.
The specific causes of juvenile myelomonocytic leukaemia are not known. However, five genes have been identified as mutated in the bone marrow cells of 90% of JMML patients. These mutations are considered to be the triggers of the disease. In the remaining 10% of patients the genetic mutation that acts as a leukaemia promoter is still unknown.
Some developmental diseases, such as neurofibromatosis type 1 and CBL syndrome, can make a child more likely to develop juvenile myelomonocytic leukaemia.
Leukaemia, like other cancers, is not contagious. See section Leukaemia, bone marrow and blood cells.
How is juvenile myelomonocytic leukaemia classified?
Juvenile myelomonocytic leukaemia is highly heterogeneous and has always been difficult to classify. Over time it has been referred to as juvenile chronic myeloid leukaemia, childhood chronic myelomonocytic leukaemia or childhood monosomy 7 syndrome. See LMMJ classification.
What are the symptoms of juvenile myelomonocytic leukaemia?
The clinical presentation of juvenile myelomonocytic leukaemia is variable and, in general, symptoms at diagnosis are due to leukaemic cells having infiltrated the bone marrow and other organs, hindering their correct functioning.
This can lead to:
- Tiredness, weakness, dizziness and paleness, as a result of low levels of red blood cells in the blood (anaemia).
- Appearance of bruises and small pink spots on the skin (petechiae), as well as other types of bleeding such as nosebleeds or gum bleeding, due to low numbers of platelets in the blood.
- Fever and infections that do not evolve well (bronchitis, tonsillitis…), due to deficient generation of immune system cells.
- Sometimes painful swelling of the lymph nodes, the liver or spleen occur, due to the accumulation of leukaemic cells.
- Pain or a feeling of fullness in the ribs
- Dry cough
- Loss of appetite and insufficient growth
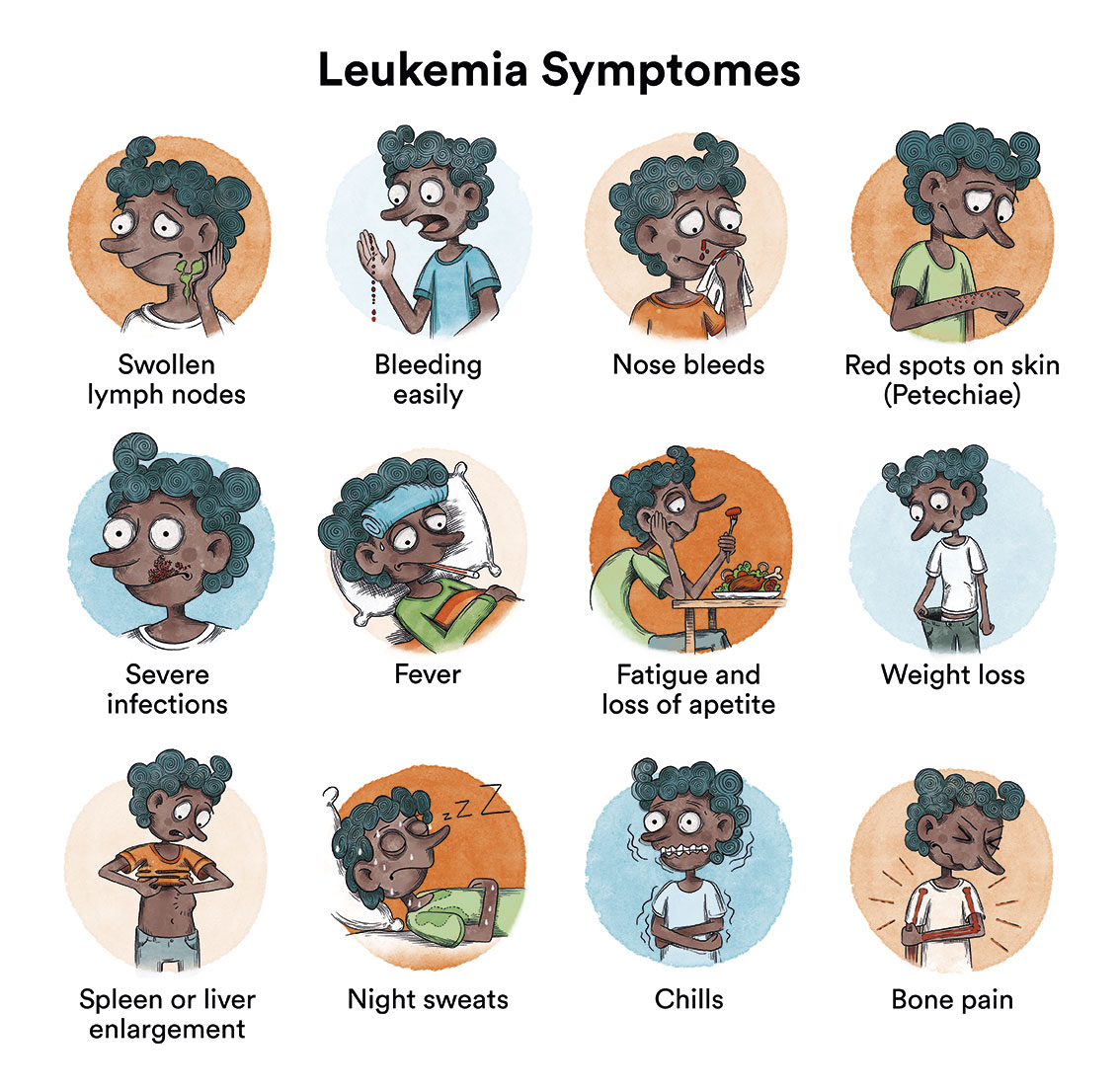
At the onset of the disease, all these symptoms can be mistaken for those of a viral infection, therefore obtaining a final diagnosis of the patient can take several weeks. However, in general, this does not affect the child’s curative options.
How is juvenile myelomonocytic leukaemia diagnosed?
In addition to the basic blood and bone marrow studies common to all types of leukaemia (morphology and cell count, immunophenotyping), cytogenetic analyses (to detect chromosomal abnormalities) and molecular analyses (to detect mutations) are essential for typing and classifying the disease. Certain genetic alterations can predict the aggressiveness of the disease and the patient’s risk of relapse. All patients with suspected juvenile myelomonocytic leukaemia undergo genetic analysis in which their tumour cells are compared with their normal cells. In more than 90% of patients with juvenile myelomonocytic leukaemia, mutations in one of these five are detected: PTPN11, KRAS, NRAS, NF1 or CBL. All these genes belong to the so-called RAS signalling pathway, which controls cell growth and division. Mutations found in juvenile myelomonocytic leukaemia cause hyperactivation of this pathway, causing tumour cells to grow uncontrollably.
What is the treatment for juvenile myelomonocytic leukaemia?
There is currently no specific treatment for juvenile myelomonocytic leukaemia. The only treatment that has proven to be curative is haematopoietic stem cell transplantation (bone marrow transplantation) from an HLA-compatible donor. Despite this, it is a therapy that is not without risks and complications, and approximately 30% of children relapse after transplantation.
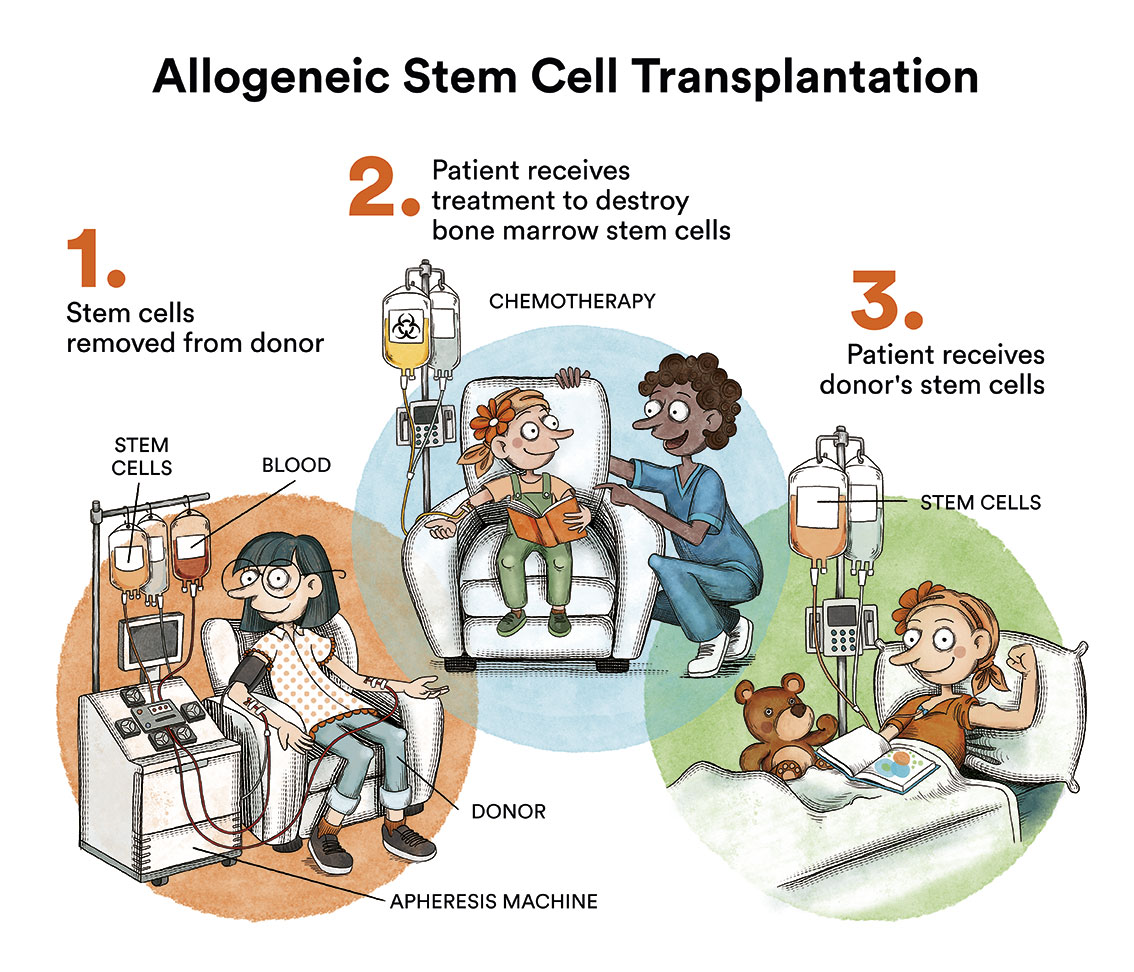
Without treatment, juvenile myelomonocytic leukaemia progresses rapidly. It is possible to use chemotherapy (the use of special drugs to kill cancer cells) to temporarily control juvenile myelomonocytic leukaemia and prepare the patient for transplantation, but not as a curative therapy. One of the treatments traditionally used prior to bone marrow transplantation is the combination of the chemotherapeutic drugs busulfan, cyclophosphamide and mephalan. However, azacitidine has recently been approved for the same purpose, and it can be administered in paediatric patients as of one month of age. Patients who achieve a temporary remission of leukaemia with these chemotherapy cycles will have a better long-term prognosis. According to the Leukemia and Lymphoma Society, transplantation is recommended for all children with mutations in the genes NF1 and PTPN11. However, the “watchful waiting” approach is adopted in patients with mutations in the CBL gene and some patients with mutations in NRAS, where disease severity is less acute and it can sometimes resolve without intervention. See LMMJ classification.

Due to the limited treatment alternatives for juvenile myelomonocytic leukaemia, there is a possibility that the responsible clinical team in charge of the child may propose the inclusion of the child in clinical trials with new research phase drugs. It is highly recommended that the child be treated at a referral centre with haematologists experienced in the treatment of this disease.
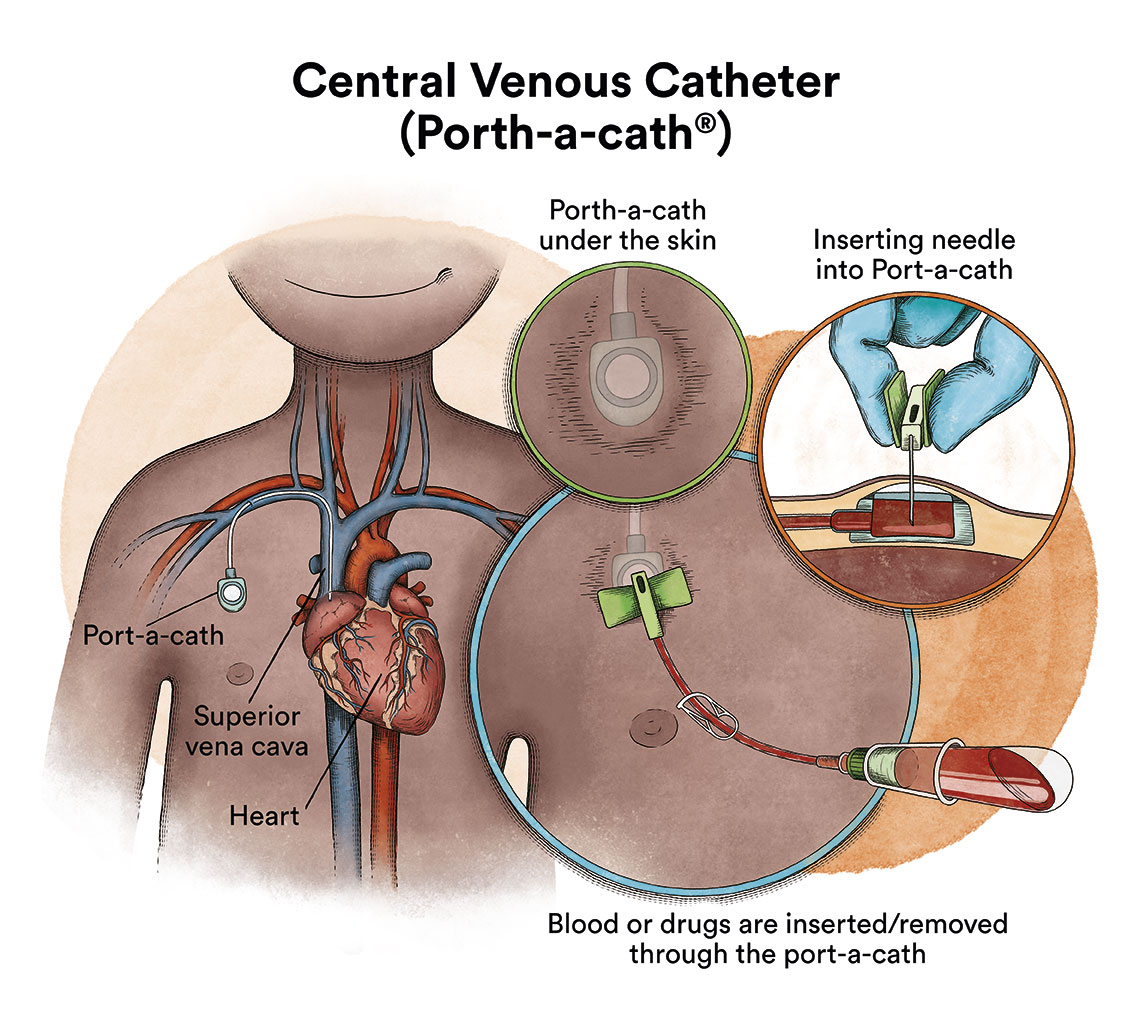
When chemotherapy is administered intravenously, to avoid repeatedly puncturing a vein, specialists implant a special device called a catheter (content in spanish). The catheter is inserted into a large vein and it allows for the administration of all types of medication, as well as for drawing blood for blood tests, thus avoiding the need for repeated needle punctures.
There is a type of catheter, called a port-a-cath, which is attached to a round plastic or metal reservoir under the skin of the chest. The port-a-cath is very practical for children because it is under the skin and does not allow the child to pull it out, it is less likely to become infected than other types of catheters and it allows the child to bathe.
What are the chances of juvenile myelomonocytic leukaemia patients being cured?
The chances of cure are determined by the characteristics of the patient, the disease (genetic/molecular alterations), the treatment given and the response to this treatment.
About 50% of patients achieve long-term complete remission after bone marrow transplantation. It should be noted that this therapy is not without complications. Between 30-40% of children with juvenile myelomonocytic leukaemia undergoing treatment and allogeneic transplantation will relapse within the first year. Sometimes a second bone marrow transplant is considered for these children.
New treatments for juvenile myelomonocytic leukaemia
JMML has historically represented a clinical challenge mainly due to the limited number of therapeutic options for treatment and the high risk of mortality in children with the most aggressive forms of the disease. Although bone marrow transplantation is currently the only curative treatment, advances in bone marrow transplantation research have led to the identification of new therapeutic strategies that are already being used in patients or explored in clinical trials:
- Hypomethylating agents: Some JMML patients with high levels of DNA methylation are associated with more aggressive forms of the disease. Methylation is a chemical modification that occurs in DNA and controls the activity of genes. To counteract this hypermethylation, azacitidine (Vidaza®) has recently been approved for the treatment of JMML patients. This drug belongs to the group of so-called hypomethylating agents, which are able to reduce DNA methylation. Possible combinations of azacitidine with other drugs are currently being studied to improve results.
- MEK inhibitors: Most patients with juvenile myelomonocytic leukaemia have mutations in genes that cause hyperactivation of the RAS signalling pathway, which controls cell proliferation, resulting in uncontrolled tumour cell growth. For this reason, blocking the RAS pathway with so-called MEK inhibitors is one of the therapeutic strategies currently under research for relapsed and/or refractory JMML. These inhibitors include drugs such as trametinib (Mekinist®).
Monitoring
After completing the treatment, the child will have regular check-ups performed by their haematologist and by other specialists where necessary. Monitoring is carried out to assess possible relapse and to follow-up and treat possible long-term complications. These checks are progressively spaced out until they are carried out once a year. Long-term follow-up is recommended at least annually in order to early detection and treatment of any sequelae arising from treatment or leukaemia, should they appear.
Links of interest concerning medical issues relating to juvenile myelomonocytic leukaemia
Juvenile myelomonocytic leukaemia (JMML). Cancer Research UK
Juvenile myelomonocytic leukaemia (JMML). The Leukaemia Foundation
Juvenile Myelomonocytic Leukemia. St Jude Children’s Research Center
Juvenile myelomonocytic leukemia (JMML). Lekemia & Lymphoma Foundation
Links of interest on other topics related to juvenile myelomonocytic leukaemia
CHILDHOOD LEUKAEMIA MATERIAL
- Babies also have leukaemia. Josep Carreras Leukaemia Foundation. (content in Spanish)
- Childhood leukaemia. The little unstoppable ones. Josep Carreras Leukaemia Foundation. (content in Spanish)
- Medulin cut-out set. Josep Carreras Leukaemia Foundation. (content in Spanish)
The Josep Carreras Foundation has a story “The tough baby” aimed at children or siblings suffering from leukaemia. It is especially aimed at children up to the age of 6. If you want to order it, please send us an e-mail to imparables@fcarreras.es.
BONE MARROW TRANSPLANT
- Bone Marrow Transplant Guide. Josep Carreras Leukaemia Foundation.
- What is HLA and how does it work? Josep Carreras Leukaemia Foundation. (content in Spanish)
- Graft-versus-Host Disease Josep Carreras Leukaemia Foundation. (content in Spanish)
- History of Bone Marrow Transplantation. Josep Carreras Leukaemia Foundation. (content in Spanish)
- How is the search for an anonymous donor conducted? Josep Carreras Leukaemia Foundation. (content in Spanish)
- Care guide for transplanted children. TransplantCHild.
- Stem cell transplantation: a colouring book. Leukaemia and Lymphoma Society.
SUPPORT MANUALS
- How to deal with leukaemia and lymphoma in children? Leukemia & Lymphoma Society.
- LIVING BY LEARNING. Action protocol for students with cancer AFANION.
- Support guide for parents of children with cancer ASION.
- A guide for young people and adolescents with cancer ASION.
- Pupils with cancer. A guide for teachers ASION.
- The importance of parental behaviour when a child has cancer ASION.
- My child has cancer. What do I do? FARO.
FOOD
- How to maintain a healthy diet during treatment? Josep Carreras Leukaemia Foundation. (content in Spanish)
- “Bon appetit”. Dietary advice during treatment AFANION.
- ‘Jabel’s magic recipes’. Isabel Rojas Murcia, Carolina Mangas Gallardo.
OTHER
- Information on the long-term and late effects of treatment for leukaemia/lymphoma in children Leukemia & Lymphoma Society.
- My sibling has cancer Josep Carreras Leukaemia Foundation. (content in Spanish)
- School in a hospital Josep Carreras Leukaemia Foundation. (content in Spanish)
- Educating illusions. Guide for psycho-educational intervention in children and adolescents with cancer FARO.
- Cancer in adolescents Josep Carreras Leukaemia Foundation. (content in Spanish)
- ‘Leukaemia and adolescents’ documentary Josep Carreras Leukaemia Foundation.
- ‘Babies also have leukaemia’ documentary Josep Carreras Leukaemia Foundation.
- 7 ways to wear a scarf Josep Carreras Leukaemia Foundation. (content in Spanish)
- ‘Princess Luzie and the chemo knights’ story ASPANAFOA.
- ‘Let’s go to chemotherapy’ story.
- ‘Let’s go to radiotherapy’ story.
- ‘Gasparín Super Quimio’ story Spanish Federation of Parents of Children with Cancer.
- ‘Charlie Brown and leukaemia’ story.
- ‘Toby and the flying machine’ story.
- ‘The fairy of the stars’ story AECC.
- ‘Lina the little swallow’ story Osakidetza.
Useful links: local entities (resources and services)
All these organisations are external to the Josep Carreras Foundation.
ANDALUCÍA
ARAGÓN
ASTURIAS
CASTILLA LA MANCHA
CASTILLA LEÓN
CATALUÑA
VALENCIAN COMMUNITY
EXTREMADURA
GALICIA
BALEARIC ISLANDS
CANARY ISLANDS
LA RIOJA
MADRID
- AAA (asociación de adolescentes y Adultos Jóvenes con Cáncer)
- ASION
- FUNDACIÓN CAICO
- FUNDACIÓN ALADINA
- FUNDACIÓN UNOENTRECIENMIL
MURCIA
NAVARRA
BASQUE COUNTRY
Support and assistance
We also invite you to follow us through our main social media (Facebook, Twitter and Instagram) where we often share testimonies of overcoming this disease.
If you live in Spain, you can also contact us by sending an e-mail to imparables@fcarreras.es so that we can help you get in touch with other people who have overcome this disease.