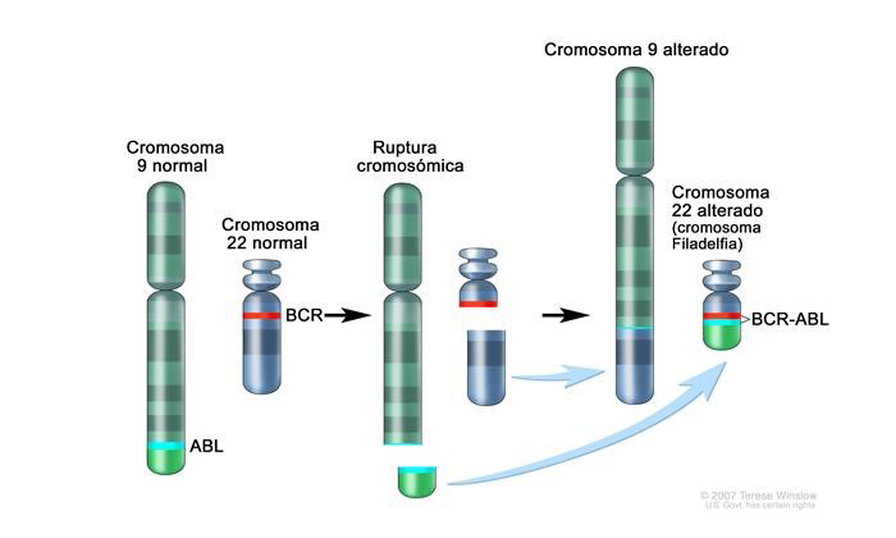
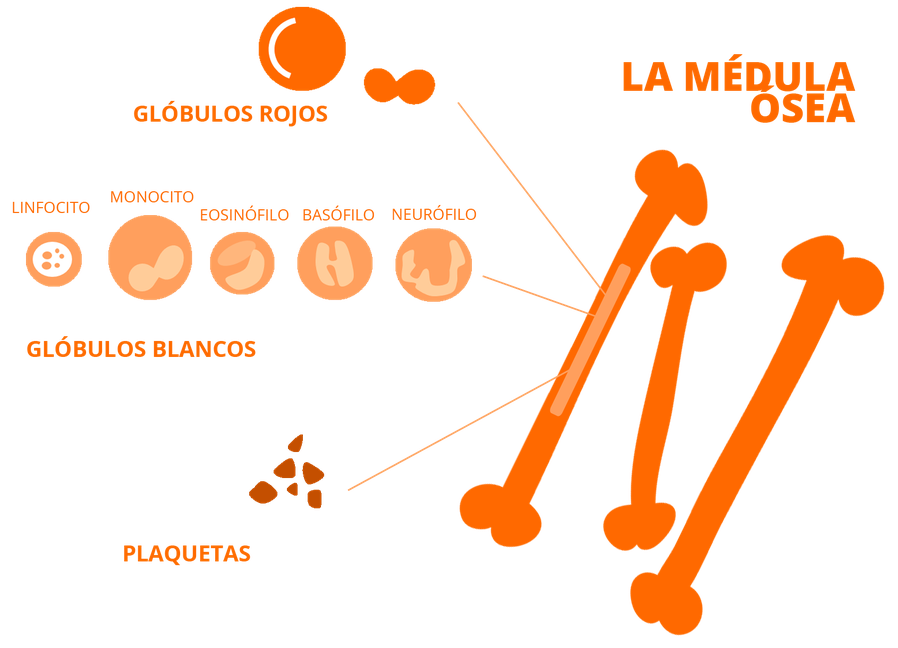
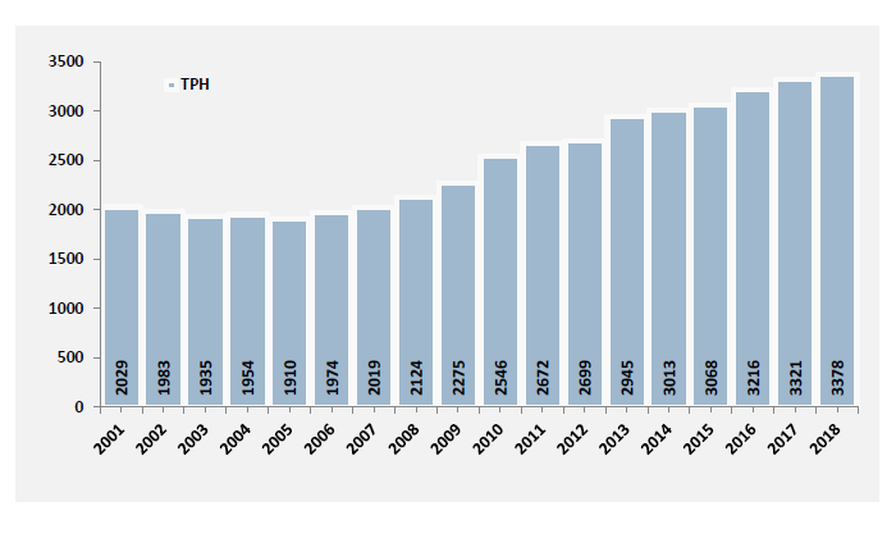
Aunque somos muchos los que luchamos cada día para ser imparables, desgraciadamente la leucemia no es aún una enfermedad curable para todos y en todos los casos. 2 de cada 10 niños no superan la enfermedad y aproximadamente el 50% de los adultos.
El vacío que deja la pérdida de un ser querido es enorme y, a menudo, se hace muy difícil continuar hacia adelante. Hacerse a la idea, aceptarlo, pensar en el futuro, … incluso realizar las acciones más cotidianas cuesta mucho después de la pérdida de una persona importante para nosotr@s.
El duelo es un proceso, no un estado
El duelo tiene distintas etapas y requiere, sobre todo, de tiempo. Normalmente, los psicólogos y psiquiatras describen el proceso de duelo mediante las siguientes fases:
1. Fase de la negación
2. Fase de la ira
3. Fase de la negociación
4. Fase de la depresión
5. Fase de aceptación
Suele ser frecuente pasar por estas etapas, pero no necesariamente debemos pasar por todas y en este orden. Además, es posible que unas se mezclen con otras o se dilaten más en el tiempo. No hay un proceso de duelo “normal”, todos somos diferentes y vivimos la vida y la muerte de forma distinta.
Y precisamente porque somos distint@s, hay quien se encierra más en sí mismo durante el duelo, y hay quien hablar y compartir experiencias le ayuda a afrontar mejor esta etapa. En muchas ciudades de España existen grupos de duelo para compartir estos momentos e incluso, en algunas asociaciones, hay grupos de duelo en función de quien era la persona que nos ha dejado: un/a herman@, un/a hij@, la pareja, un/a amig@, etc.
Algunos grupos de duelo son conducidos por terapeutas profesionales y otros por personas que han sufrido una pérdida y, voluntariamente, una vez superado el proceso de duelo, deciden utilizar su experiencia para ayudar a otras personas. En todos los casos se practica la escucha activa y respetuosa y en algunos, además, se complementa con técnicas de respiración o meditación.
A continuación, podéis encontrar algunas referencias de grupos de duelo en España*:
Organizaciones que están en varias ciudades de España y recursos online:
● Asociación Española contra el Cáncer
● Grupos de duelo Renacer en España: en Asturias, Barcelona, Vilanova i la Geltrú (Barcelona), Girona, Lloret de Mar (Girona), Córdoba, Galicia, San Sebastián, La Bañeza (León), Tafalla (Navarra), Pamplona, La Rioja, Dos Hermanas (Sevilla) y Zaragoza.
● Asociación Alma y Vida (Padres y madres en duelo): en Sevilla, Algeciras (Cádiz), Puertollano (Ciudad Real), Chiclana (Cádiz), Jaén y Córdoba.
● Apoyo en red (on line)
● Fundación Mario Losantos del Campo
Andalucía:
● Grupo de duelo Emucesa (Granada)
● Asociación Alma y Vida (Padres y madres en duelo): en Sevilla, Algeciras (Cádiz), Chiclana (Cádiz), Jaén y Córdoba.
Aragón:
● ASPANOA (atención y apoyo en el duelo en casos de niños con cáncer)
● Grupo de apoyo al duelo Monzón (Monzón)
● Asociación Aragonesa Madres y mujeres Arco Iris
● Grupo de duelo Renacer (Zaragoza)
Asturias:
● Asociación Galbán (atención y apoyo en el duelo en casos de niños con cáncer)
Baleares:
● ASPANOB (atención y apoyo en el duelo en casos de niños con cáncer)
● Asociación de ayuda al duelo Decir Adiós (Ibiza y Formentera)
● Associació Lligams (Menorca)
Catalunya:
● AFANOC (atención y apoyo en el duelo en casos de niños con cáncer)
● Associació Suport i Companyia (Barcelona) (atención y apoyo en el duelo en casos de pacientes hematológicos)
● Fundació Roses contra el Càncer (Roses)
● Oncovallès (Vallès Oriental)
● Villassar de Dalt contra el Càncer
● Grups de dol AVES (Barcelona)
● Servei de Suport al Dol (Girona)
● Associació Ca n’Eva (Terrassa)
● Grup d’acompanyament al dol de Lleida (Lleida)
● Lliga contra el Càncer de Tarragona
● Grup de Suport al dol de Sabadell
● Renèixer Girona (para padres que han perdido un hijo)
Castilla la Mancha:
● AFANION (atención y apoyo en el duelo en casos de niños con cáncer)
● Duelo Albacete Talitha (Albacete)
● Asociación Alma y Vida Puertollano (Ciudad Real)
Castilla y León:
● PYFANO (atención y apoyo en el duelo en casos de niños con cáncer)
● Grupos de duelo Renacer La Bañeza (León)
Comunidad Valenciana:
● ASPANION (atención y apoyo en el duelo en casos de niños con cáncer)
● Asociación Valenciana de Apoyo al Duelo Caminar (Valencia)
● Associació Petjada (Castelló)
Extremadura
Islas Canarias
● ACODUEL (Las Palmas de Gran Canaria)
● Grupo de Duelo ‘Para siempre en el corazón’ (Tenerife)
● Asociación Acompañar (Lanzarote)
● Grupo de ayuda mutua para padres y madres en duelo (Gran Canaria)
● Grupo de ayuda mutua para personas en duelo por la muerte de un ser querido (La Palma)
La Rioja
● FARO (grupo de duelo para padres de niños con cáncer)
Madrid:
● Fundación Aladina (grupo de duelo para padres de niños con cáncer)
● Asociación de mutua ayuda ante el duelo
● Fundación Mario Losantos del Campo
● Centro de escucha San Camilo
Murcia:
Navarra
● Asociación Goizargi (Pamplona)
● Grupos de duelo Renacer (Tafalla)
País Vasco:
● Juneren Egoak (grupo de duelo para padres de niños con cáncer en Guipúzcoa)
● Izargi (San Sebastián)
● Asociación Krisalida de Ayuda al duelo (Bilbao)
● Grupo de duelo Renacer (San Sebastián)
● Bidegin
* La Fundación ha recopilado estos datos con el fin de ofrecer direcciones de utilidad para las personas que están buscando un acompañamiento a su proceso de duelo. No conocemos a tod@s las entidades. Esta lista constituye simplemente un recurso meramente informativo.