Información avalada por ![]()
¿Qué es la Anemia de Fanconi y a quién afecta?
La Anemia de Fanconi es una enfermedad hereditaria de muy baja frecuencia entre la población. A pesar de su nombre, Suele caracterizarse por otras muchas manifestaciones además de la anemia.
Las más relevantes:
- Insuficiencia medular progresiva (anemia, leucopenia y trombocitopenia).
- Malformaciones congénitas
- Alta predisposición tumoral, especialmente a desarrollar leucemias mieloblásticas agudas (LMA), síndromes mielodisplásicos (SMD) y tumores sólidos.
 Su frecuencia es de 1 cada 300.000. Se trata de una enfermedad genética recesiva en la mayoría de los casos. Por lo tanto, para que un individuo padezca esta enfermedad, las dos copias de los genes involucrados en la Anemia de Fanconi (la heredada del padre y la heredada de la madre) han de estar afectadas. La afección generalmente se diagnostica en niños entre los 3 y 14 años de edad. Cuando los genes están implicados en una predispocisicón tumoral en portadores, es importante realizar un seguimiento adecuado en unidades especializadas. Es fundamental recibir un adecuado consejo genético de los familiares en riesgo, especialmente en parejas portadoras.
Su frecuencia es de 1 cada 300.000. Se trata de una enfermedad genética recesiva en la mayoría de los casos. Por lo tanto, para que un individuo padezca esta enfermedad, las dos copias de los genes involucrados en la Anemia de Fanconi (la heredada del padre y la heredada de la madre) han de estar afectadas. La afección generalmente se diagnostica en niños entre los 3 y 14 años de edad. Cuando los genes están implicados en una predispocisicón tumoral en portadores, es importante realizar un seguimiento adecuado en unidades especializadas. Es fundamental recibir un adecuado consejo genético de los familiares en riesgo, especialmente en parejas portadoras.
Consejo genético
Al tratarse en la mayoría de los casos de mutaciones recesivas, para que un individuo paadezca Anemia de Fanconi, los dos alelos de un determinado gen FANC heredados de sus padres han de poseer la mutación patogénica.
Si un hombre y una mujer, ambos portadores de mutaciones en un determinado gen FANC tienen descendencia, 3 de cada 4 hijos, en promedio, serán sanos (en promedio, 2 serán portadores y 1 será completamente normal: no tendrá afectado ninguno de los dos alelos de dicho gen).
No obstante, en promedio, 1 de cada 4 hijos poseerá mutaciones en ambos alelos, por lo que padecerá la enfermedad con una severidad difícil de definir con antelación. En el caso del gen FANCB, al estar en el cromosoma X, todos los afectados son hombres y solo las madres son portadoras. En estas familias es muy importante detectar mujeres portadoras en edad reproductiva debido al alto riesgo de engendrar un hijo varón afecto.
Los individuos que poseen una copia funcional y la otra mutada se definen como portadores de la enfermedad. Salvo en el caso de portadores con una mutación en los genes FANCD1/BRCA2, FANCS/BRCA1, FANCN/PALB2 o FANCJ/BRIP1, que afectan a una minoría de los pacientes, no existe ninguna evidencia de que portadores de AF posean riesgos de padecer enfermedades con mayor frecuencia que el resto de la población.
Otras consideraciones dentro del consejo genético (planificación familiar, diagnóstico prenatal o diagnóstico preimplantacional):
Es importante contactar con especialistas para recibir consejo genético que ayudara a una persona o una familia a entender el riesgo de padecer una enfermedad genética, educar a las personas sobre esa enfermedad y evaluar el riesgo de transmitir la enfermedad a sus hijos, para tener una adecuada planificación familiar y un correcto diagnóstico prenatal o preimplantacional.
Frecuentemente el asesor genético trabaja con familias para identificar a las personas en riesgo. De ser apropiado o necesario, hablará de test genéticos, interpretará resultados y supervisará todos los análisis adicionales, ayudas u opciones de investigación al alcance de los miembros de la familia. Los asesores genéticos trabajan como parte de un equipo de salud, junto con médicos especializados, trabajadores sociales, enfermeras, genetistas clínicos y otros especialistas para ayudar a las familias a tomar decisiones informadas sobre su salud. También serán fuente de ayuda y apoyo en momentos difíciles
Si desea contactar con los especialistas que le puedan ayudar en el diagnóstico, seguimiento y tratamiento de pacientes con anemia de Fanconi puede hacerlo de la siguiente manera:
Fundación Anemia de Fanconi España
secretariaFAF@anemiadefanconi.org
654 617 808
¿Cuáles son las causas de la Anemia de Fanconi?
La Anemia de Fanconi es una enfermedad compleja desde el punto de vista genético, ya que hasta el momento se han descrito 22 genes involucrados en esta enfermedad. La falta de función de cualquiera de estos 22 genes produce la enfermedad. Estos genes anormales dañan las células, lo cual les impide reparar el ADN dañado. Esta enfermedad se debe principalmente a mutaciones en los genes, existiendo algunas correlaciones entre subtipos de Anemia de Fanconi, mutaciones genéticas y el comportamiento de la enfermedad.
En los pacientes con mutaciones de FANC C la aplasia es más precoz y la supervivencia más corta que en los portadores de mutaciones en FANC A y FANC G.
Los pacientes con FANC G tienen mayor riesgo de desarrollar leucemias y tumores sólidos que los del tipo FANC A.
Los subtipos FANC D1 y FANC N conllevan riesgo de desarrollar meduloblastomas o tumores de Wilms. Las mutaciones heterocigotas en FANC D1, FANC J y FANC N comportan un riesgo incrementado de cáncer de mama.
¿Cuáles son los síntomas de la Anemia de Fanconi?
 Aproximadamente el 90% de los niños y niñas con Anemia de Fanconi tienen alteraciones de la función de la médula ósea que conducen a una anemia aplásica. Los individuos afectados padecen cansancio y debilidad debido a la anemia, infecciones recurrentes debido a la neutropenia y problemas de coagulación debido a la trombocitopenia.
Aproximadamente el 90% de los niños y niñas con Anemia de Fanconi tienen alteraciones de la función de la médula ósea que conducen a una anemia aplásica. Los individuos afectados padecen cansancio y debilidad debido a la anemia, infecciones recurrentes debido a la neutropenia y problemas de coagulación debido a la trombocitopenia.
Las características clínicas asociadas a la Anemia de Fanconi son el retraso del crecimiento pre y postnatal, malformaciones renales, gastrointestinales, genitourinarias, cardíacas y esqueléticas, cabeza, ojos y boca pequeños, hipogonadismo, sordera parcial, anormalidades cutáneas como híper o hipopigmentación y manchas “café con leche” y elevados niveles de a-fetoproteína en sangre. Tres de cada diez pacientes, sin embargo, no presentan malformación alguna.
Los problemas hematológicos suelen aparecer en la edad escolar, alrededor de los 7 años, aunque esto es muy variable. Las alteraciones hematológicas afectan a la mayor parte de los pacientes de Anemia de Fanconi antes de los 40 años. El 90% de los casos se diagnostican antes de la adolescencia. Los pacientes muestran recuentos anormalmente bajos de células sanguíneas, tanto de glóbulos rojos (anemia), glóbulos blancos (leucopenia) y de plaquetas (trombopenia). La primera manifestación suele ser una trombopenia aislada en más de la mitad de los casos, observándose petequias o hematomas, o episodios de sangrado nasal o gastrointestinal. Posteriormente se hacen evidentes los signos de anemia que incluyen principalmente palidez, astenia y anorexia. La tendencia a padecer procesos infecciosos (secundaria a la deficiencia de glóbulos blancos) suele ser de aparición tardía. Una vez iniciada la afectación hematológica, la evolución suele conducir a la pancitopenia (anemia + leucopenia + trombopenia) en un periodo de tiempo extraordinariamente variable.
¿Cómo se diagnostica la Anemia de Fanconi?
- Diagnóstico clínico
El diagnóstico basado en observaciones clínicas puede resultar extremadamente dificultoso debido a la gran variabilidad de síntomas que muestran los pacientes de Anemia de Fanconi. Este hecho, unido a que un 30% de los pacientes AF no muestran malformación alguna, supone que muchos pacientes de Anemia de Fanconi sólo acuden al médico cuando los síntomas de la enfermedad hematológica son manifiestos. La marcada reducción de una o más series sanguíneas suele poner al especialista ante la sospecha de una anemia hereditaria, que debe ser confirmada mediante estudios clínicos adicionales, así como mediante análisis citogenéticos y moleculares.
- Diagnóstico genético
La Anemia de Fanconi es una enfermedad compleja desde el punto de vista genético, ya que hasta el momento se han descrito 22 genes involucrados en esta enfermedad: FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI, FANCJ/BRIP1/BACH1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4/BTBD12, FANCQ/ERCC4/XPF, FANCR/RAD51, FANCS/BRCA1, FANCT/UBE2T, FANCU/XRCC2, FANCV/REV7,FANCW/RFWD3
La confirmación diagnóstica de la enfermedad se realiza mediante ensayos de fragilidad cromosómica (roturas de cromosomas) inducidas por agentes que generan enlaces cruzados en las cadenas de ADN. Estos estudios suelen realizarse sobre linfocitos de sangre periférica o fibroblastos de piel. Estos ensayos deben realizarse por laboratorios experimentados en el chequeo de enfermedades genéticas. La Red Nacional de Investigación en Anemia de Fanconi ofrece a los pacientes españoles la realización de confirmaciones diagnósticas de la enfermedad, así como entrenamiento a laboratorios que deseen especializarse en estos ensayos. En aproximadamente un 20% de los pacientes de Anemia de Fanconi, la confirmación diagnóstica de la enfermedad puede ser particularmente compleja debido a un fenómeno conocido como mosaicismo somático. El mosaicismo somático deriva del hecho de que algunas células de la sangre pueden corregir, por diferentes mecanismos, la mutación del gen causante de la enfermedad, por lo que en la sangre del paciente cohabitan células de Anemia de Fanconi y células sanas.
El diagnóstico del subtipo genético se realiza con técnicas de secuenciación (Sanger o NGS) y MLPA. Mediante el estudio mutacional se pueden identificar portadores de la enfermedad, y realizar estudios de diagnóstico prenatal o preimplantacional.
- Diagnóstico diferencial
Es importante que el equipo tenga experiencia en el manejo de los pacientes de Anemia de Fanconi. Hay algunas enfermedades que pueden confundirse en el diagnóstico. A tener en cuenta:
- Disqueratosis congénita
- Anemia de Blacfan-Diamond
- Síndrome de Shwachman-Diamond
- Neutropenia congénita severa
- Síndrome TAR (trombocitopenia con ausencia de radios)
- Trombocitopenia amegacariocítica
- Síndrome de Baller-Gerold
- Síndrome de rotura de Hijmegen
- Síndrome de Rothmund-Thomson
- Síndrome de Roberts
- Síndrome de rotura de Varsovia
- Focomelia DK
- Síndrome de hidrocefalia VACTERL
- Síndrome de Wiskott-ALdrich
¿Cuál es el tratamiento de la Anemia de Fanconi?
Las transfusiones de hematíes y plaquetas se hacen necesarias cuando la anemia y trombocitopenia son intensas y sintomáticas.

Los derivados androgénicos (oximetolona a dosis de 2–4 mg/kg/día) pueden disminuir el grado de anemia y ahorrar transfusiones, pero debe vigilarse la aparición de efectos secundarios (virilización, aceleración del crecimiento, disfunción hepática y tumores hepáticos). Su empleo debe ser transitorio a la espera de un trasplante de progenitores hematopoyéticos (trasplante de médula ósea).
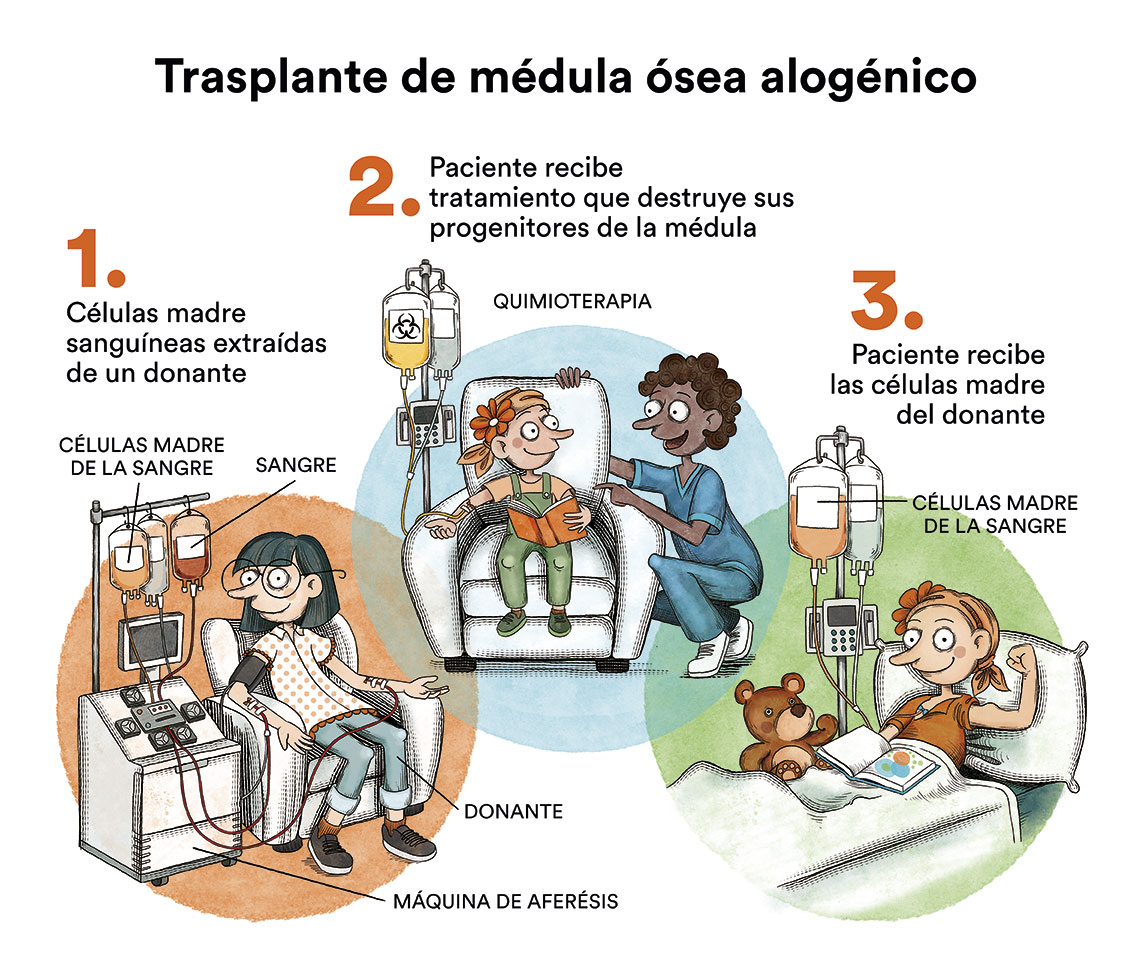
El trasplante de progenitores hematopoyéticos es el único tratamiento que puede restaurar una hematopoyesis normal, pero no revierte las lesiones somáticas ni previene el desarrollo de tumores sólidos. EL donante ideal es un hermano HLA idéntico, pero, actualmente, si esta opción no existe, los trasplantes de donantes voluntarios HLA compatibles también son muy buenos.
El empleo de los clásicos regímenes mieloablativos con ciclofosfamida e irradiación dan lugar a malos resultados por la hipersensibilidad de las células de la Anemia de Fanconi a los agentes genotóxicos que producen graves lesiones en los tejidos. Es por ello que deben emplearse regímenes de acondicionamiento de intensidad reducida adaptados a estos pacientes y evitar siempre la radioterapia.
Los pacientes con Anemia de Fanconi receptores de un trasplante de médula ósea requieren un seguimiento estrecho por el riesgo de desarrollar neoplasias, que incluye revisiones de crecimiento y endocrinológicos, citogenética en médula ósea y exámenes orales regulares con biopsia ante cualquier lesión sospechosa.
¿Qué probabilidades tienen de curarse los niños Anemia de Fanconi?

La probabilidad de transformación de la enfermedad en una leucemia mieloide aguda es del 30% y la incidencia de tumores sólidos del 28% a los 40 años.
Sin trasplante de médula ósea, la mediana de supervivencia de los pacientes con Anemia de Fanconi es de 24 años. La incidencia de insuficiencia medular llega a ser del 90% a los 40 años. La muerte, en el 90% de los casos, se debe a las alteraciones hematológicas (aplasia medular, síndromes mielodisplásicos o leucemia mieloide aguda) o a complicaciones derivadas de su tratamiento.
Los trasplantes realizados a partir de un hermano compatible y sano ofrecen una probabilidad de supervivencia libre de enfermedad hematológica de hasta el 90%. El empleo de un donante HLA anónimo no es óptimo, pero va mejorándose cada día. EL pronóstico asociado al trasplante es mejor si el paciente ha recibido pocas transfusiones, si el paciente es joven y si no ha desarrollado un proceso leucémico. Por tanto, en el caso de que el paciente posea un donante adecuado, es recomendable realizar el trasplante antes de que comience a requerir soporte transfusional, o cuando los estudios citogenéticos en la médula ósea sugieran el inicio de una mielodisplasia. De ahí la importancia del correcto seguimiento de estos pacientes a nivel de su sangre periférica y médula ósea. La Red Nacional de Investigación en Anemia de Fanconi ha estandarizado protocolos para el trasplante de estos pacientes de acuerdo a los mejores estándares internacionales, y ofrece información a los centros especializados que soliciten información al respecto. Contactar con la Fundación Anemia de Fanconi.
Seguimiento
- Seguimiento hematológico
Los pacientes con Anemia de Fanconi tienen un elevado riesgo de presentar anemia aplásica, síndromes mielodisplásicos o leucemia mieloide aguda. La probabilidad de transformación en LMA es del 30% y la incidencia de tumores sólidos del 28% a los 40 años. Es muy importante realizar un control riguroso de sus parámetros hematológicos. A modo orientativo, los controles recomendados son los siguientes:
- Hemograma mediante muestra de sangre periférica cada 3-4 meses
- Mielograma, citogenética, análisis CD34+ y contenido CFCs) mediante aspirado de médula ósea cada 1 año-1,5 años si no se observan cambios significativos.
El seguimiento hematológico del paciente ha de comenzarse desde el diagnóstico de su enfermedad.
- Seguimiento y prevención de tumores de cabeza y cuello
Los pacientes con Anemia de Fanconi tienen una predisposición, mucho más elevada que el resto de la población, a desarrollar tumores sólidos. Por ello el seguimiento por especialistas
que conozcan la enfermedad es de vital importancia. Hay un riesgo acumulado de padecer algún tipo de tumor a 40 años de un 25%. No hay casos descritos en pacientes menores de 10 años. En pacientes trasplantados el riesgo aumenta a partir del 5º año post-trasplante. Las recomendaciones para revisiones y seguimiento rutinario son las siguientes:
- Pacientes no trasplantados: desde los 10 años de edad
- Pacientes trasplantados: desde el momento del trasplante independientemente de su edad
Debe establecerse vigilancia de la cavidad bucal, nasofaringe, orofaringe, hipofaringe y laringe cada semestre si no hay cambios significativos. En caso de que haya cambios, debe vigilarse lichenplanus, leucoplaquia y eritroplaquia mediante una biopsia cada 2-3 meses. Y si hay lesión sospechosa de carcinoma, tomar inmediatamente una biopsia y realizar seguimiento cada 2-3 meses, además de realizar una radiografía anual.
- Seguimiento y prevención de tumores ginecológicos
Debido al riesgo de cáncer de cérvix, vagina y vulva, debe comenzarse con el seguimiento ginecológico a partir de los 16 años o de la primera menstruación. De la misma forma, se debe realizar un seguimiento de control de mamas también a partir de la primera menstruación o de los 20 años.
EL seguimiento debe consistir anualmente en: un frotis Papanicolau, examen meticuloso del tracto genital inferior, seguimiento endocrino y test del virus del Papiloma.
- Seguimiento y cuidados dentales
Se recomienda iniciar las revisiones dentales a la edad de un año y medio y realizar 2 revisiones al año. Se recomienda poner en contacto al dentista que trata a un paciente Anemia de Fanconi con su médico especialista, con objeto de que le informe sobre las posibles complicaciones asociadas a intervenciones bucales.
Se recomienda revisar lesiones persistentes, ulceraciones sospechosas o leucoplaquias y ponerse en contacto con el especialista por si fuera conveniente realizar una biopsia del tejido bucal. Se recomienda también ponerse en contacto con el especialista, en el caso de sangrado continuado, o pérdidas dentales sin causa aparente.
En el caso de que el paciente presente niveles anormalmente bajos de plaquetas o glóbulos blancos, conviene informar de ello al dentista, para prevenir hemorragias o infecciones en el caso de que el paciente vaya a ser sometido a una intervención dental.
Enlaces de interés sobre temas médicos relacionados con la Anemia de Fanconi
- Guía básica para el diagnóstico. Fundación Anemia de Fanconi España
- Seguimiento de pacientes de Anemia de Fanconi. Fundación Anemia de Fanconi España
Enlaces de interés sobre otros temas relacionados con la Anemia de Fanconi
MATERIALES LEUCEMIA INFANTIL
- Los bebés también tienen leucemia. Fundación Josep Carreras contra la leucemia.
- Leucemia infantil. Los pequeños imparables. Fundación Josep Carrerascontra la leucemia.
- Juego recortable Medulín. Fundación Josep Carreras contra la leucemia.
La Fundación Josep Carreras dispone de un cuento “El bebé forzudo” dirigido a niños o hermanos que padecen leucemia. Está especialmente dirigido a niños hasta los 6 años. Si quieres solicitarlo, puedes enviarnos un correo a imparables@fcarreras.es.
TRASPLANTE DE MÉDULA ÓSEA
- Guía del Trasplante de Médula Ósea. Fundación Josep Carreras contra la leucemia.
- ¿Qué es el HLA y cómo funciona? Fundación Josep Carreras contra la leucemia.
- La Enfermedad Injerto contra Receptor Fundación Josep Carreras contra la leucemia.
- Historia del Trasplante de Médula Ósea. Fundación Josep Carreras contra la leucemia.
- ¿Cómo se realiza la búsqueda de un donante compatible anónimo? Fundación Josep Carreras contra la leucemia.
- Guía de cuidados para niños trasplantados. TransplantCHild.
- El trasplante de células madre: un libro para colorear. Leukeamia and Lymphoma Society.
MANUALES DE APOYO
- ¿Cómo enfrentarse a la leucemia y el linfoma en niños? Leukemia & Lymphoma Society.
- VIVIR APRENDIENDO. Protocolo de actuación para alumnos con cáncer AFANION.
- Guía de apoyo para padres de niños oncológicos ASION.
- Guía para jóvenes y adolescentes con cáncer ASION.
- Alumnado con cáncer. guía para docentes ASION.
- La importancia del comportamiento de los padres cuando un niño tiene cáncer ASION.
- Mi hijo tiene cáncer. ¿Qué hago? FARO.
ALIMENTACIÓN
- ¿Cómo mantener una alimentación saludable durante el tratamiento? Fundación Josep Carreras contra la leucemia.
- “Buen provecho”. Consejos dietéticos durante el tratamiento AFANION.
- ‘Las recetas mágicas de Jabel’. Isabel Rojas Murcia, Carolina Mangas Gallardo.
OTROS
- Información sobre los efectos a largo plazo y tardíos del tratamiento para la leucemia o el linfoma en los niños Leukemia & Lymphoma Society.
- Mi hermano tiene cáncer Fundación Josep Carreras contra la leucemia.
- La escuela en un hospital Fundación Josep Carreras contra la leucemia.
- Educando ilusiones. Guía para la intervención psicoeducativa en niños y adolescentes con cáncer FARO.
- El cáncer en la adolescencia Fundación Josep Carreras contra la leucemia.
- Documental ‘La leucemia y los adolescentes’ Fundación Josep Carreras contra la leucemia.
- Documental ‘Los bebés también tienen leucemia’ Fundación Josep Carreras contra la leucemia.
- 7 formas de ponerse un pañuelo Fundación Josep Carreras contra la leucemia.
- Cuento ‘La princesa Luzie y los caballeros de la quimio’ ASPANAFOA.
- Cuento ‘Vamos a quimioterapia’.
- Cuento ‘Vamos a radioterapia’.
- Cuento ‘Gasparín Super Quimio’ Federación Española de Padres de Niños con Cáncer.
- Vídeo ‘Charlie Brown y la leucemia’.
- Cuento ‘Toby y la máquina voladora’.
- Cuento ‘El hada de las estrellas’ AECC.
- Cuento ‘Lina la pequeña golondrina’ Osakidetza.
Enlaces de interés: entidades locales (recursos y servicios)
Todas estas organizaciones son externas a la Fundación Josep Carreras.
ANDALUCÍA ARAGÓN ASTURIAS CASTILLA LA MANCHA CASTILLA LEÓN CATALUÑA COMUNIDAD VALENCIANA EXTREMADURA GALICIA ISLAS BALEARES ISLAS CANARIAS LA RIOJA MADRID MURCIA NAVARRA PAÍS VASCO
ANDALUCÍA ARAGÓN ASTURIAS CASTILLA LA MANCHA CASTILLA LEÓN CATALUÑA COMUNIDAD VALENCIANA EXTREMADURA GALICIA ISLAS BALEARES ISLAS CANARIAS LA RIOJA MADRID MURCIA NAVARRA PAÍS VASCO
Apoyo y ayuda
Te invitamos también a seguirnos a través de nuestras redes sociales principales (Facebook, Twitter e Instagram) en las que, a menudo, compartimos testimonios de superación.
Si resides en España, también puedes ponerte en contacto con nosotros enviándonos un correo electrónico a imparables@fcarreras.es para que te ayudemos a ponerte en contacto con otras familias que han superado esta enfermedad.