Información avalada por ![]()
¿Cómo funciona la médula ósea y cuáles son los tipos de células sanguíneas?
Los síndromes mielodisplásicos son un tipo de cáncer de las células de la sangre y de la médula ósea.
Ver apartado Leucemia, médula ósea y células sanguíneas
¿Qué son los síndromes mielodisplásicos?
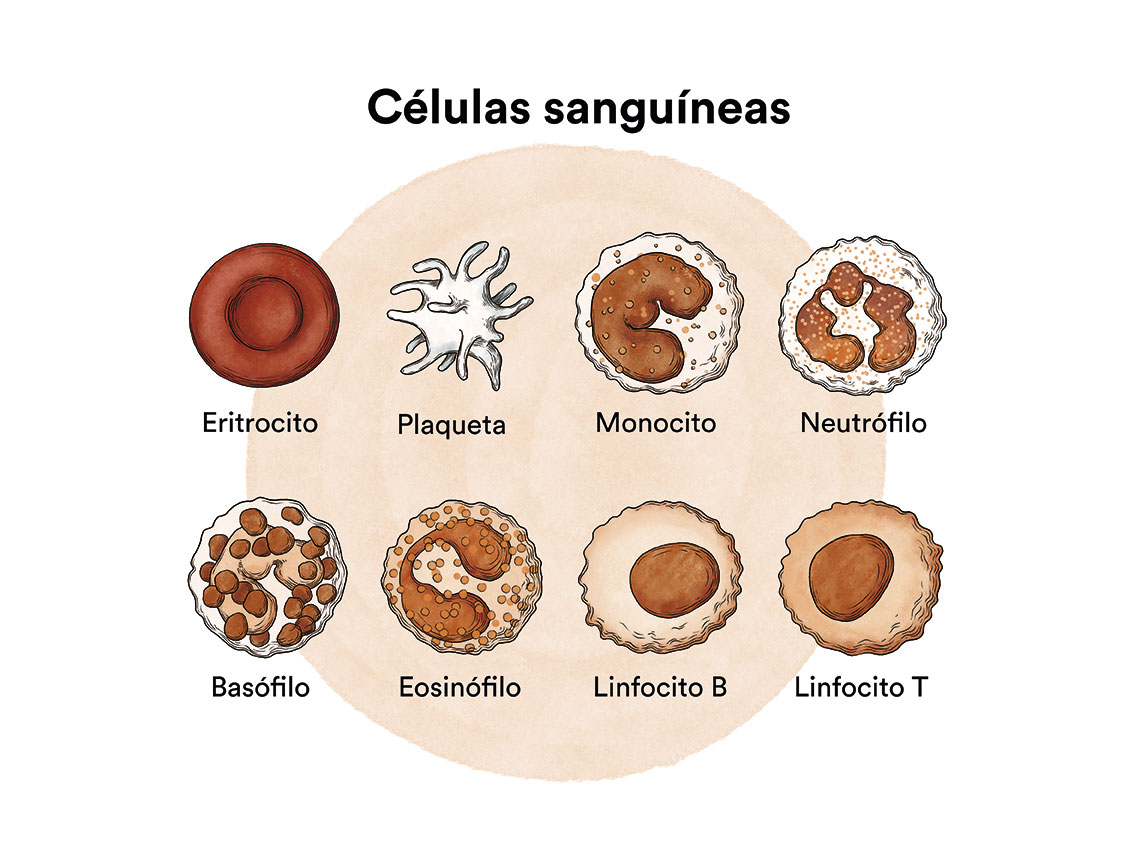
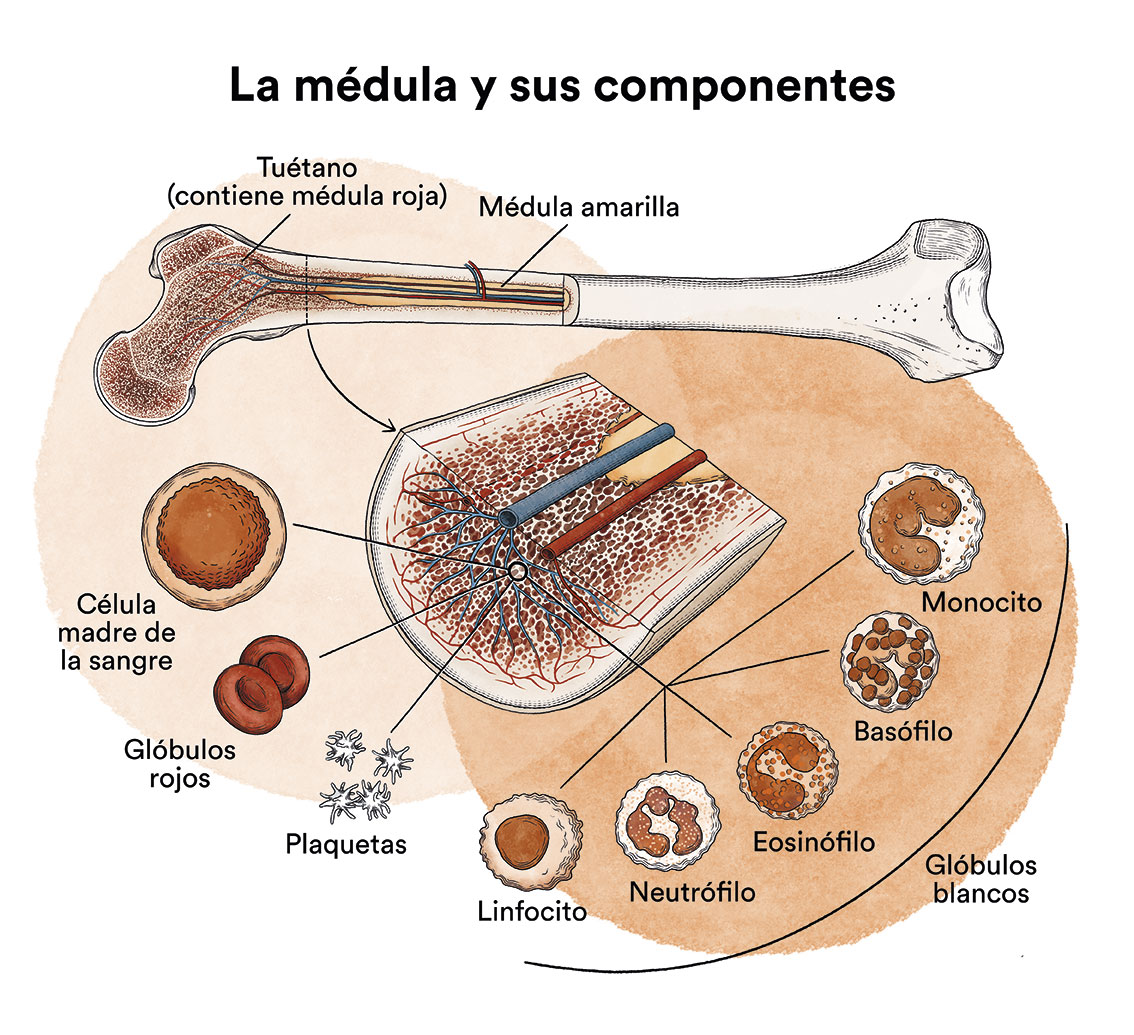
Los síndromes mielodisplásicos (SMD) son un grupo de enfermedades de la médula ósea, órgano encargado de fabricar las células de la sangre (glóbulos rojos, glóbulos blancos y plaquetas). En condiciones normales estas células se reproducen y maduran en la médula ósea hasta salir y circular por la sangre. En los síndromes mielodisplásicos (SMD) la médula ósea fabrica estas células de forma anómala, tanto en número como en maduración o funcionamiento, y estas anomalías se detectan en la sangre cuando hacemos una analítica (hemograma) y miramos la sangre al microscopio.
En los síndromes mielodisplásicos suele haber una cifra disminuida de algunas de las células de la sangre con algunas alteraciones morfológicas (displásicas). En un paciente de síndrome mielodisplásico, las células madre sanguíneas (células inmaduras) en la médula ósea, no se convierten en glóbulos rojos, glóbulos blancos o plaquetas maduras. Estas células sanguíneas inmaduras, llamadas blastocitos, no funcionan como debieran y mueren en la médula ósea o poco después de entrar en la sangre. Esto deja menos espacio para que se formen glóbulos blancos, glóbulos rojos y plaquetas sanas en la médula ósea. Cuando hay menos células sanguíneas sanas, se pueden presentar infecciones, anemia o sangrado fácil.
Asimismo, en los pacientes de síndromes mielodisplásicos hay alteraciones del ADN que afectan cromosomas, genes (mutaciones) o la metilación (modificaciones del ADN que pueden desactivar la función de un gen) y también de otras estructuras relacionadas con el ADN.
¿Cuáles son los diferentes tipos de síndromes mielodisplásicos?
Los SMD forman un grupo de enfermedades heterogéneo con pronósticos y tratamientos muy distintos. La Organización Mundial de la Salud (OMS) clasifica los SMD en función del porcentaje de células precursoras en la médula ósea que muestran displasia (se ven anormales al microscopio), de qué porcentaje de glóbulos rojos primitivos son sideroblastos en anillo (células que contienen anillos de depósitos de hierro alrededor de los núcleos), qué porcentaje de blastos hay en la médula ósea o en la sangre (células sanguíneas muy inmaduras) y ciertas alteraciones cromosómicas. Esta clasificación distingue los siguientes subtipos de SMD:
1. Síndrome mielodisplásico con displasia de una sola línea (MDS-SLD)
Se observa displasia en al menos 10% de 1 tipo de células precursoras de glóbulos rojos, glóbulos blancos y/o megacariocitos (las células que producen plaquetas) en la médula ósea.
La persona tiene recuentos bajos de un o 2 tipos de células sanguíneas, pero un número normal de otro(s) tipo(s).
Existe un número normal (menos de 5%) de células muy primitivas llamadas blastos en la médula ósea. Además, hay muy pocos blastos (o no hay ninguno) en la sangre.
Es raro que este tipo de síndrome mielodisplásico progrese a una leucemia mieloide aguda (LMA). Los pacientes con este tipo de síndrome mielodisplásico pueden a veces vivir mucho tiempo, incluso sin recibir tratamiento. En la clasificación anterior, OMS-2008 se denominaba como anemia refractaria (RA), neutropenia refractaria (RN) y trombocitopenia refractaria (RT), dependiendo del tipo de célula afectado.
2. Síndrome mielodisplásico con displasia multilínea (MDS-MLD)
Se observa displasia en por lo menos 10% en 2 o 3 tipos de células precursoras de glóbulos rojos, glóbulos blancos y/o megacariocitos (las células que producen plaquetas) en la médula ósea.
La persona tiene recuentos bajos de al menos un tipo de célula sanguínea.
Existe un número normal (menos de 5%) de células inmaduras, llamadas blastos, en la médula ósea. Además, hay muy pocos blastos (o no hay ninguno) en la sangre.
Este es el tipo de MDS en el pasado, según la clasificación OMS-2008, se denominaba citopenia refractaria con displasia multilínea (RCMD).
Si los glóbulos rojos están afectados, pueden tener una cantidad adicional de hierro. La citopenia resistente al tratamiento puede progresar y volverse leucemia mieloide aguda (LMA).
3. Síndrome mielodisplásico con sideroblastos en anillo (MDS-RS)
En este tipo de síndrome mielodisplásico, muchas de las células precursoras de los glóbulos rojos son sideroblastos en anillo (células que contienen anillos de depósitos de hierro alrededor de los núcleos). Para este diagnóstico, al menos el 15% de los glóbulos rojos primitivo (eritroblastos) tienen que ser sideroblastos en anillo (o al menos el 5% si las células también tienen una mutación en el gen SF3B1).
Esta afección se subdivide en dos tipos, basándose en cuántos de los tipos de células en la médula ósea se ven afectados por la displasia (anormalidad de las células):
- Síndrome mielodisplásico con sideroblastos en anillo con displasia unilinaje (MDS-RS-SLD): displasia en un solo tipo de célula.
- Síndrome mielodisplásico con sideroblastos en anillo con displasia multilinaje (MDS-RS-MLD): displasia en más de un tipo de célula.
Se trata de un subtipo de síndrome mielodisplásico poco frecuente. Rara vez se transforma a una leucemia mieloide aguda y, en general, el pronóstico es mejor que en otros tipos de SMD. Anteriormente, en la clasificación de la OMS-2008 se conocía este subtipo como anemia refractaria con sideroblastos en anillo (RARS).
4. Síndrome mielodisplásico con exceso de blastos (MDS-EB)
En este subtipo hay más blastos (células sanguíneas inmaduras) de lo normal en la médula ósea y/o en la sangre. El paciente también presenta recuentos sanguíneos bajos en, al menos, un tipo de célula sanguínea. Puede o no haber displasia grave en la médula ósea.
Esta afección se subdivide en dos tipos, basándose en qué porcentaje de la médula ósea son blastos:
- Síndrome mielodisplásico con exceso de blastos (MDS-EB1): los blastos constituyen del 5% al 9% de las células en la médula ósea, o del 5% al 19% de las células en sangre.
- Síndrome mielodisplásico con exceso de blastos (MDS-EB2): los blastos constituyen del 10% al 19% de las células en la médula ósea, o del 2% al 4% de las células en sangre.
Este subtipo representa alrededor del 25% de todos los diagnósticos de síndromes mielodisplásicos. Es uno de los tipos que más probabilidades tiene de convertirse en una leucemia mieloide aguda, especialmente en el subtipo MDS-EB2). Anteriormente, en la clasificación de la OMS-2008 se conocía este subtipo como anemia refractaria con exceso de blastos (RAEB).
5. Síndrome mielodisplásico con del aislada del cromosoma 5 (Síndrome 5q-)
En este tipo de síndrome mielodisplásico, a los cromosomas les falta una parte del cromosoma 5. Puede haber, además, otra anomalía cromosómica, siempre y cuando no sea la pérdida de todo o parte del cromosoma 7). La cantidad de blastos es normal.
La persona también presenta recuentos bajos de uno o 2 tipos de células sanguíneas (generalmente glóbulos rojos), y existe displasia en al menos un tipo de célula en la médula ósea.
Este tipo de síndrome mielodisplásico es poco frecuente y ocurre más a menudo en mujeres de edad avanzada. Por razones que no están claras, los pacientes con este tipo de síndrome mielodisplásico suelen tener un pronóstico favorable. A menudo, viven por mucho tiempo y rara vez padecen leucemia mieloide aguda.
6. Síndrome mielodisplásico inclasificable (MSD-U)
En esta categoría se incluyen los síndromes mielodisplásicos poco frecuentes. Algunos pueden tener alteraciones citogenéticas típicas de otros SMD sin que haya displasia ni alteraciones relevantes de las células sanguíneas.
Existen otros dos tipos de síndromes mielodisplásicos a tener en cuenta que no están incluidos en la clasificación de la Organización Mundial de la Salud. Se trata de:
- Los síndromes mielodisplásicos secundarios: aquellos que se desencadenen en personas que hay recibido previamente tratamiento de quimioterapia o radioterapia por otro tipo de cáncer. Las personas con este tipo de SMD suelen tener un peor pronóstico que los SMD habituales.
- El síndrome mielodisplásico hipoplásico: En este tipo se observan pocas células madre sanguíneas en la médula ósea. En este caso, las defensas del organismo enfermo atacan las células madre de la médula ósea haciendo que muchas sean eliminadas. Como este tipo hipoplásico de SMD tiene un mecanismo especial, su tratamiento también lo es. Se suele utilizar medicamentos que reduzcan la potencia de nuestras defensas, los llamados “inmunosupresores”.
¿Cuáles son las causas de los síndromes mielodisplásicos?
Se desconoce por qué aparecen los síndromes mielodisplásicos, pero en la mayoría de los casos, son enfermedades adquiridas relacionadas con el envejecimiento o son debidas a la exposición ambiental, laboral o no, a sustancias tóxicas o tratamientos como la radioterapia y/o quimioterapia, entre otros.
Los síndromes mielodisplásicos, como otros tipos de cáncer, no son contagiosos. Ver apartado Leucemia, médula ósea y células sanguíneas.
¿Cuáles son los síntomas de los síndromes mielodisplásicos?
Es frecuente que en las etapas iniciales de los síndromes mielodisplásicos los pacientes no noten ninguna molestia. En estos casos, habitualmente el síndrome mielodisplásico se descubre tras unos resultados de recuentos bajos de células sanguíneas en una analítica de control o para otros procesos médicos.
En los casos en los que el paciente no se encuentra bien y acude a su médico, los síntomas y su gravedad dependerá del tipo de células afectadas y de cuan bajos sean los recuentos sanguíneos.
Si hay anemia por disminución de los glóbulos rojos es frecuente el cansancio y la debilidad (cuando es grave se puede notar mareo, palpitaciones, sudor…). En casos más graves puede haber síntomas derivados de la disminución de glóbulos blancos y/o plaquetas que provocan infecciones y/o hemorragias, respectivamente.
Cuando predominan las alteraciones proliferativas hay riesgo de trombosis ya que el aumento de glóbulos rojos y/o plaquetas puede obturar los vasos sanguíneos, especialmente si la persona fuma, tiene colesterol o hipertensión arterial.
Además, dado que la médula ósea trabaja a mayor ritmo de lo normal, el hígado y el bazo pueden aumentar de tamaño para que también contribuyan a esta proliferación; este aumento de tamaño puede dar molestias en la barriga (hinchazón, dolor, estreñimiento …).
También puede haber síntomas como cansancio, falta de apetito, pérdida de peso, picor (es lo que llaman síntomas constitucionales).
¿Cómo se diagnostican los síndromes mielodisplásicos?
Además de los estudios básicos en sangre y medula ósea (morfología, recuento, inmunofenotipo), los estudios citogenéticos (para detectar anomalías cromosómicas concretas) y estudios moleculares (para detectar alteraciones genéticas especificas) son fundamentales para tipificar y clasificar la enfermedad. Determinadas alteraciones genéticas y moleculares se correlacionan con la sensibilidad al tratamiento y el pronóstico.
En definitiva, para diagnosticar un SMD se debe estudiar:
1) Analítica con estudio microscópico de las células de la sangre, donde veremos alteraciones del número y forma de las células;
2) Aspirado de la médula ósea: observaremos que las células predecesoras de las de la sangre están en mucha más cantidad de lo normal y, en ellas, hay alteraciones de forma y tamaño; observaremos un porcentaje variable de células inmaduras anómalas (blastos) con posibles alteraciones en sus cromosomas y genes; y, por último, observaremos o no sideroblastos en anillo; además con el aspirado se realizan estudios citogenéticos y moleculares que son claves para determinar el riesgo y pronóstico de la enfermedad.
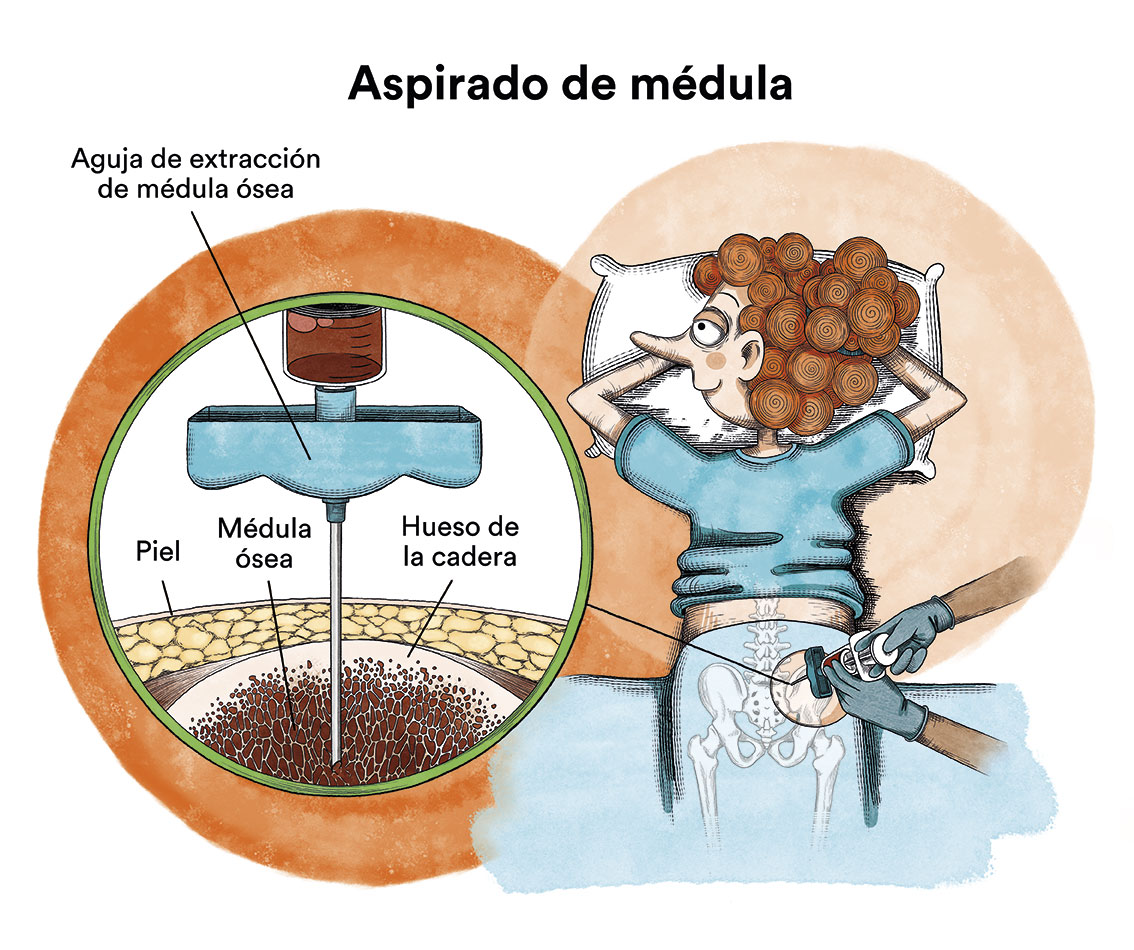
3) Biopsia de la médula ósea: esta se realiza para confirmar el diagnóstico y, en ella, veremos un aumento considerable del número de células en la médula ósea y podemos encontrar un grado leve-moderado de fibrosis (una especie de «red o tela» que dificulta el normal funcionamiento de la misma).
¿Cuál es el tratamiento de los síndromes mielodisplásicos?
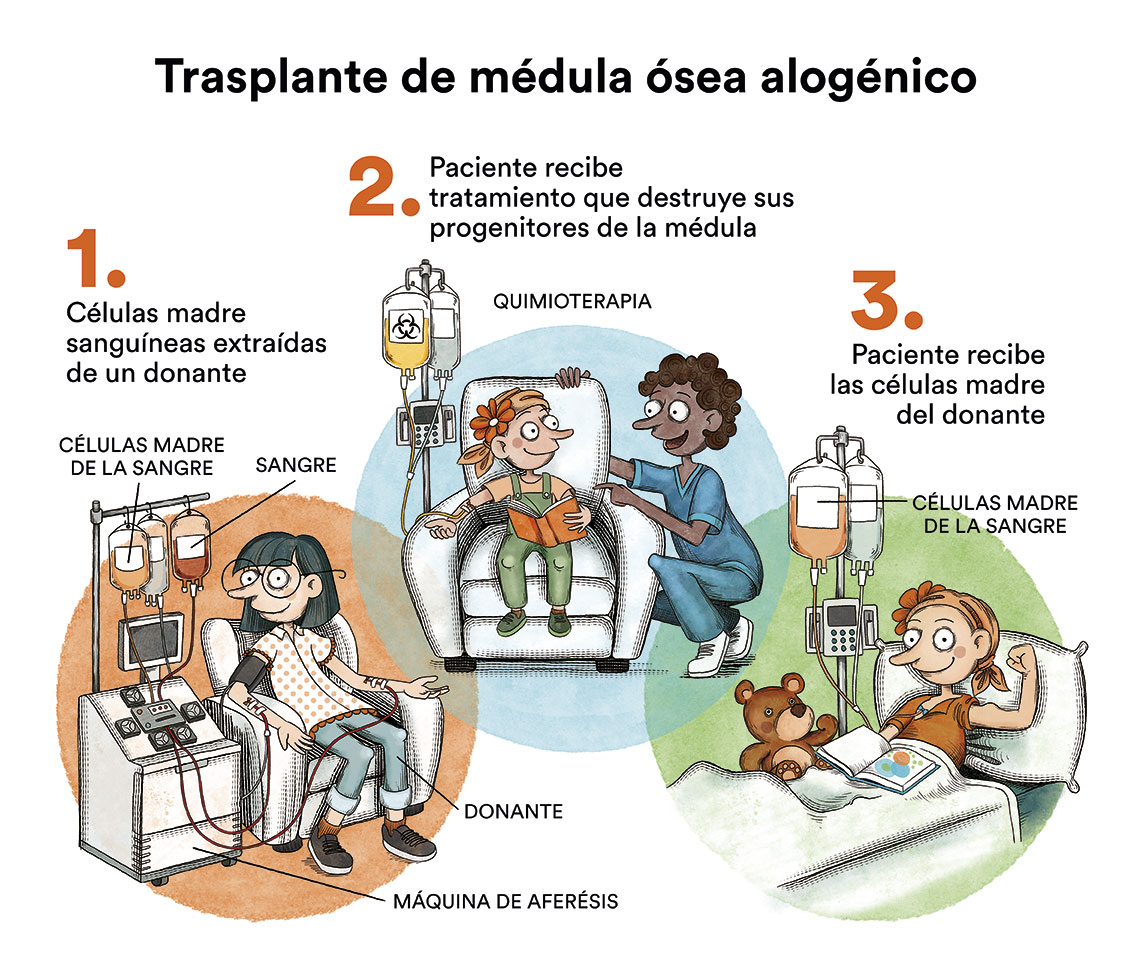
Como se ha comentado anteriormente, existen muchos tipos de síndromes mielodisplásicos con características y tratamientos muy distintos. El único tratamiento curativo sería un trasplante de médula ósea alogénico (de donante) pero por sus riesgos, solo se reserva a SMD de alto riesgo de transformarse a una leucemia mieloide aguda y en pacientes jóvenes.
Para establecer el nivel de riesgo de cada uno de los síndromes mielodisplásicos se utiliza un sistema pronóstico consensuado internacionalmente que está en constante revisión. Dicho de otro modo, es un sistema que pretende dilucidar con mayor precisión si la enfermedad será más o menos agresiva. Este índice se llama IPSS-R y se ha realizado a partir de los datos de miles de pacientes a nivel internacional. Este índice se basa en:
- el número de blastos en la médula ósea
- tipo de células sanguíneas están alteradas (glóbulos rojos, plaquetas y neutrófilos)
- si las células de la médula ósea presentan cromosomas alterados.
En las siguientes tablas se puede ver una versión detallada de este sistema:
Sistema Pronóstico Internacional Revisado (IPSS-R)
| Variable | 0 | 0.5 | 1 | 1.5 | 2 | 3 | 4 |
| Citogénica | Muy buena | – | Buena | – | Intermedia | Pobre | Muy Pobre |
| Blastos en médula ósea | Menos de 2% | – | 3 ó 4% | – | Entre 5 y 10% | Más de un 10% | – |
| Nivel de Hemoglobina (en gramos por decilitro) | Más de 10 | – | Menos de 10 y más de 8 | Menos de 8 | – | – | – |
| Cantidad de plaquetas por microlitro de sangre | Más de 100.000 | Entre 50.000 y 100.000 | Menos de 50.000 | – | – | – | – |
| Cantidad de neutrófilos por microlitro de sangre | Más de 800 | Menos de 800</ | – | – | – | – | – |
El sistema pronóstico IPSS-R divide las alteraciones de los cromosomas en cinco grupos según su riesgo:
- Muy buenas: los varones que pierden el cromosoma Y o los casos en los que se pierde un fragmento del cromosoma 11.
- Buena: los casos con citogenética normal o con una sola alteración consistente en la pérdida de un segmento del cromosoma 5, del 12 o del 20. También los casos en que haya dos alteraciones si una de ellas es del 5.
- Intermedia: si hay una sola alteración consistente en la pérdida de un segmento del 7, o la aparición de un tercer cromosoma 8, o de un tercer cromosoma 19. También la aparición de un cromosoma 17 alterado.
- Pobre: perder por completo el cromosoma 7, alteraciones en el cromosoma 3, los casos con tres alteraciones juntas o dos si una de ellas es una pérdida parcial o completa del cromosoma 7.
- Muy pobre: los casos llamados complejos, en los que hay más de tres alteraciones a la vez.
Dependiendo de la puntuación total que se sumen todos estos factores, el riesgo según el IPSS-R será:
Grupos pronósticos del Sistema Pronóstico Internacional Revisado (IPSS-R)
| Grupos de riesgo | Puntos totales | Mediana de supervivencia en años (sin recibir tratamiento) | Mediana de años hasta que 1 de cada 4 pacientes en este grupo desarrollen una leucemia aguda (sin recibir tratamiento) |
| Riesgo muy bajo | Menos de 1,5 | 8,8 | No se ha alcanzado |
| Riesgo bajo | Más de 1,5 y menos de 3 | 5,5 | 10,8 |
| Riesgo intermedio | Más de 3 y menos de 4,5 | 3 | 3,2 |
| Riesgo alto | Más de 4,5 y menos de 6 | 1,6 | 1,4 |
| Riesgo muy alto | Más de 6 | 0,8 | 0,7 |
Para los síndromes mielodisplásicos actualmente tenemos pocos tratamientos eficaces y no hay un esquema de tratamiento definido. Lo que sí hay son unos criterios de respuesta al tratamiento específicos de estas enfermedades. En general se tiene en cuenta qué características (displásicas o proliferativas) predominan en cada paciente. Por ello, se plantean distintas opciones de tratamiento:
- Si el descenso de los niveles de células sanguíneas es leve y el tipo de SMD de bajo riesgo, es suficiente intentar controlar los síntomas que tiene el paciente. Lo más razonable puede ser “vigilar y esperar”, y no optar por tratamiento o transfusiones. En estos casos, se controlan los niveles sanguíneos mediante análisis periódicas y, si existe algún cambio, se harían más pruebas para valorarlo. Este tipo de aproximación terapéutica genera mucha incertidumbre en los pacientes y a menudo hay sensación de “pasividad” ante la situación por parte del equipo médico. Pero está demostrado que, de momento, esta es la mejor opción para algunos tipos de SMD. La única opción curativa para un SMD es el trasplante de médula ósea alogénico. Sin embargo, esta terapia tiene un riesgo vital importante y secuelas así hay que reservarla para pacientes cuyo riesgo es alto.
- En pacientes jóvenes (<70 años), sin otras patologías graves, con riesgo moderado o alto de que el SMD se transforme a una leucemia aguda, el tratamiento de elección debe ser curar la enfermedad mediante un trasplante de médula ósea alogénico (de donante), que es el único tratamiento curativo, aunque puede haber complicaciones graves derivadas de infecciones y/o rechazo de la médula ósea trasplantada. En estas ocasiones se plantea la utilización de diversas quimioterapias de administración oral y/o intravenosa
- En pacientes de riesgo moderado o alto en que no se puede realizar un trasplante de médula ósea, ya sea por edad o por otras patologías graves no compatibles, lo más sensato es realizar las transfusiones de sangre cuando se necesite e intentar retrasar la progresión de la enfermedad con fármacos. En estos casos ni la curación ni la modificación de la evolución natural de la enfermedad son posibles.
Transfusiones sanguíneas (terapia de soporte)
Muchos de los pacientes con síndromes mielodisplásicos son muy dependientes de las transfusiones de sangre debido a la disminución de ciertas células sanguíneas. Estas transfusiones se suelen realizar en el hospital de día sin ingreso. Las transfusiones de hematíes o glóbulos rojos son habituales en algunos pacientes. Sin embargo, en ocasiones también puede ser necesario recibir una transfusión de plaquetas o factores estimulantes de los glóbulos blancos (G-CSF).

Los efectos secundarios del tratamiento con transfusiones de sangre son poco comunes, pero pueden observarse reacciones a la transfusión, infecciones, desarrollo de anticuerpos contra los glóbulos rojos o las plaquetas y sobrecarga de hierro en diferentes órganos del cuerpo.
Nuevos tratamientos: tratamientos modificadores de la evolución de la enfermedad
Cuando la curación del síndrome mielodisplásico no es posible por no poderse someter al paciente a un trasplante de médula ósea, existen fármacos que pueden modificar la evolución de la enfermedad con tal de mejorar la función de la médula ósea. Algunos de ellos son:
- Azacitidina (Vidaza®): se utiliza habitualmente para evitar la dependencia a las transfusiones sanguíneas, para evitar sangrados y/o infecciones. La azacitidina pertenece a una clase de medicamentos llamados agentes desmetiladores. Actúa ayudando a la médula ósea a producir glóbulos rojos normales y destruyendo las células anormales presentes en ella. Aparte del trasplante de médula ósea, la azacitidina es el primer tratamiento que ha demostrado retrasar la progresión a leucemia aguda y prolongar la vida de los pacientes con SMD de alto riesgo. Y no menos importante, también ha demostrado mejorar la calidad de vida de los pacientes tratados.
- Lenalidomida (Revlimid®): es un fármaco que se usa en algunos síndromes mielodisplásicos de bajo riesgo si tienen anemia y una lesión en el cromosoma 5 (5q-). La lenalidomida pertenece a una clase de medicamentos llamados agentes inmunomoduladores. Funciona al ayudar a la médula ósea a producir glóbulos normales y al matar las células anormales en la médula ósea.
¿Qué probabilidades tienen de curarse los pacientes con síndromes mielodisplásicos?
Como se ha comentado anteriormente, el pronóstico de los pacientes afectados de síndromes mielodisplásicos varia dependiendo de su tipo y grado (ver tipos de SMD). De todas formas, el único tratamiento para curar esta enfermedad es el trasplante de médula ósea que se reserva para pacientes jóvenes con SMD de alto riesgo (IPSS-R muy alto, alto y la mayoría de los intermedios sobre todo con una puntuación superior a 3.4, factores moleculares de mal pronóstico). También se considera en pacientes jóvenes con alta dependencia transfusional que no responden a terapias previas.
Enlaces de interés sobre temas médicos relacionados con los síndromes mielodisplásicos
- Guía para pacientes con síndrome mielodisplásico y cuidadores. GESMD (Grupo Español de Síndromes Mielodisplásicos)
- Buscador de ensayos clínicos activos. GESMD (Grupo Español de Síndromes Mielodisplásicos)
Enlaces de interés sobre otros temas relacionados con los síndromes mielodisplásicos
MATERIALES TESTIMONIALES
Puedes solicitarnos los libritos en formato papel para envío gratuito en España a través del email: imparables@fcarreras.es
TRASPLANTE DE MÉDULA ÓSEA
- Guía del Trasplante de Médula Ósea. Fundación Josep Carreras
- ¿Qué es el HLA y cómo funciona? Fundación Josep Carreras
- La Enfermedad Injerto contra Receptor. Fundación Josep Carreras
- Historia del Trasplante de Médula Ósea. Fundación Josep Carreras
- ¿Cómo se realiza la búsqueda de un donante compatible anónimo? Fundación Josep Carreras
ALIMENTACIÓN
- ¿Cómo mantener una alimentación saludable durante el tratamiento? Fundación Josep Carreras
- Guía de nutrición. Leukemia & Lymphoma Society
OTROS
- Ideas sobre qué llevarme a una cámara de aislamiento. Fundación Josep Carreras contra la leucemia
- Consejos de viaje para personas con cáncer. Fundación Josep Carreras contra la leucemia
- Manual de fisioterapia en pacientes hematológicos y trasplantados. Fundación Josep Carreras contra la leucemia
- Prevención y tratamiento de la mucositis oral. Fundación Josep Carreras contra la leucemia
- La higiene bucodental en el paciente onco-hematológico. Fundación Josep Carreras contra la leucemia
- Manual fertilidad: Padecer un cáncer de la sangre y ser padre o madre. Fundación Josep Carreras contra la leucemia
- El cuidado de la piel en el paciente onco-hematológico. Fundación Josep Carreras contra la leucemia
- Manual Estética Oncológica. Fundación Josep Carreras contra la leucemia
- Leucemia y sexualidad. Fundación Josep Carreras contra la leucemia
- 7 formas de ponerse un pañuelo. Fundación Josep Carreras contra la leucemia
Enlaces de interés: entidades locales/provinciales o estatales que pueden proveerte de recursos y servicios especializados en leucemia o en pacientes oncológicos:
En España existe un gran tejido asociativo para pacientes con cáncer hematológico que, en muchos casos, puede informarte, asesorarte e incluso, realizar algunos trámites. Estos son los contactos de algunas de ellas por Comunidades Autónomas:
Todas estas organizaciones son externas a la Fundación Josep Carreras.
ESTATAL
- CEMMP (Comunidad Española de Pacientes de Mieloma Múltiple)
- AEAL (ASOCIACIÓN ESPAÑOLA DE AFECTADOS POR LINFOMA, MIELOMA y LEUCEMIA)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana o llamando al 900 100 036 (24h).
- AELCLES (Agrupación Española contra la Leucemia y Enfermedades de la Sangre)
- FUNDACIÓN JOSEP CARRERAS CONTRA LA LEUCEMIA
- FUNDACIÓN SANDRA IBARRA
- GEPAC (GRUPO ESPAÑOL DE PACIENTES CON CÁNCER)
- MPN España (Asociación de Afectados Por Neoplasias Mieloproliferativas Crónicas)
ANDALUCÍA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ALUSVI (ASOCIACIÓN LUCHA Y SONRÍE POR LA VIDA). Sevilla
- APOLEU (ASOCIACIÓN DE APOYO A PACIENTES Y FAMILIARES DE LEUCEMIA). Cádiz
ARAGÓN
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ASPHER (ASOCIACIÓN DE PACIENTES DE ENFERMEDADES HEMATOLÓGICAS RARAS DE ARAGÓN)
- DONA MÉDULA ARAGÓN
ASTURIAS
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ASTHEHA (ASOCIACIÓN DE TRASPLANTADOS HEMATOPOYÉTICOS Y ENFERMOS HEMATOLÓGICOS DE ASTURIAS)
CANTABRIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
CASTILLA LA MANCHA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
CASTILLA LEÓN
- ABACES (ASOCIACIÓN BERCIANA DE AYUDA CONTRA LAS ENFERMEDADES DE LA SANGRE)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ALCLES (ASOCIACIÓN LEONESA CON LAS ENFERMEDADES DE LA SANGRE). León.
- ASCOL (ASOCIACIÓN CONTRA LA LEUCEMIA Y ENFERMEDADES DE LA SANGRE). Salamanca.
CATALUÑA
- ASSOCIACIÓ FÈNIX. Solsona
- FECEC (FEDERACIÓ CATALANA D’ENTITATS CONTRA EL CÁNCER
- FUNDACIÓ KÁLIDA. Barcelona
- FUNDACIÓ ROSES CONTRA EL CÀNCER. Roses
- LLIGA CONTRA EL CÀNCER COMARQUES DE TARRAGONA I TERRES DE L’EBRE. Tarragona
- MielomaCAT
- ONCOLLIGA BARCELONA. Barcelona
- ONCOLLIGA GIRONA. Girona
- ONCOLLIGA COMARQUES DE LLEIDA. Lleida
- ONCOVALLÈS. Vallès Oriental
- OSONA CONTRA EL CÀNCER. Osona
- SUPORT I COMPANYIA. Barcelona
- VILASSAR DE DALT CONTRA EL CÀNCER. Vilassar de Dalt
COMUNIDAD VALENCIANA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ASLEUVAL (ASOCIACIÓN DE PACIENTES DE LEUCEMIA, LINFOMA, MIELOMA Y OTRAS ENFERMEDADES DE LA SANGRE DE VALENCIA)
EXTREMADURA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- AFAL (AYUDA A FAMILIAS AFECTADAS DE LEUCEMIAS, LINFOMAS; MIELOMAS Y APLASIAS)
- AOEX (ASOCIACIÓN ONCOLÓGICA EXTREMEÑA)
GALICIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ASOTRAME (ASOCIACIÓN GALLEGA DE AFECTADOS POR TRASPLANTES MEDULARES)
ISLAS BALEARES
- ADAA (ASSOCIACIÓ D’AJUDA A L’ACOMPANYAMENT DEL MALALT DE LES ILLES BALEARS)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
ISLAS CANARIAS
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- AFOL (ASOCIACIÓN DE FAMILIAS ONCOHEMATOLÓGICAS DE LANZAROTE)
- FUNDACIÓN ALEJANDRO DA SILVA
LA RIOJA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
MADRID
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- AEAL (ASOCIACIÓN ESPAÑOLA DE LEUCEMIA Y LINFOMA)
- CRIS CONTRA EL CÁNCER
- FUNDACIÓN LEUCEMIA Y LINFOMA
MURCIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
NAVARRA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
PAÍS VASCO
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- PAUSOZ-PAUSO. Bilbao
CIUDADES AUTÓNOMAS DE CEUTA Y MELILLAS
- AECC CEUTA (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER)
- AECC MELILLA (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER)
Apoyo y ayuda
Te invitamos también a seguirnos a través de nuestras redes sociales principales (Facebook, Twitter e Instagram) en las que, a menudo, compartimos testimonios de superación.
Si resides en España, también puedes ponerte en contacto con nosotros enviándonos un correo electrónico a imparables@fcarreras.es para que te ayudemos a ponerte en contacto con otras familias que han superado esta enfermedad.